1. INTRODUCTION
Predicting multi-particle behavior is an extremely complicated task. The Ostwald ripening of single-phase precipitates has been extensively investigated using both analytical [1-8] and numerical methods [8-15]. The role of the coarsened phase volume fraction has been explored [3,5,6,9,10] and morphological deviations from the spherical (circular) coarsened phases at high volume fractions have already been observed in previous studies [6,9,10,15]. Ardell evaluated the role of the coarsened phase fraction using a theoretical approach and he found that the peak of the particle size distribution decreased as the particle fraction increased [3]. Brasilsford and Wynblatt [7] also found that the particle size distribution peak position decreased as the volume fraction increased, and they claimed that the effect was much less sensitive to volume fraction than proposed by Ardell [3]. Although the microstructural evolutions driven by curvature minimization have been studied extensively for many decades, current understanding of Ostwald ripening remains limited. Because we have no analytical solutions for the diffusion equations in most cases and ripening is extremely complex, multi-particle behaviors are usually predicted based on the mean-field assumption [1-8]. or numerical approaches [6,8-10,15]. Previous simulations of particle coarsening have predicted the behaviors of single phases using phenomenological models [9,10,15]. In this study, we performed a simulation of multiphase precipitates using a CALPHAD-type database to predict the behaviors of realistic precipitates. The major elements of low-Cu reactor pressure vessel (RPV) steel are Fe, Mn, Ni and Si [16-18]; therefore, we selected the Fe–Mn–Ni–Si (hereafter FMNS) quaternary system to investigate multi-phase coarsening, using the FMNS database. The phase-field method has been extensively applied to investigate the microstructural evolution of low-alloy Fe-based steel [19-21] assuming only one precipitate phase within the system. T3, T6, and T7 Mn–Ni–Si-rich (MNS) precipitates are generally observed in RPV steel [16]; therefore, we assumed the presence of three types of MNS precipitates in the system. We adopted the Kim–Kim–Suzuki (KKS) model [22] to perform multi-phase and multi-component phase-field modeling. The FMNS system, a prototype of RPV steel, is of great interest in nuclear engineering applications [16-18,23,24]. Because MNS precipitates are among the main sources of late-stage hardening for FMNS [16-18,23,24], we examined the stability of the MNS precipitates. The T3, T6, and T7 precipitate phases, major types of MNS precipitates, were considered in the UW1 database [16] in this study; we also simulated the evolution of pairs of T3–T3, T7–T7, and T3–T7 precipitates to quantify the interactions between particles.
2. UW1 CALPHAD DATABASE
In the UW1 database [16], one bcc phase representing the matrix and 12 MNS precipitate phases are considered. In our study, we selected the matrix phase and three MNS precipitate phases for simplicity. The thermodynamic parameters we used were taken from the supplementary material of ref. [16] as follows:
for bcc FMNS phase,
for the T3: Mn6⁄29Ni16⁄29Si7⁄29 phase,
for the T6: Mn1/3(Ni,Si)2/3 phase,
and for the T7: Mn1⁄2Ni1⁄3Si1⁄6 phase,
3. PHASE-FIELD MODEL AND INPUT PARAMETER EVALUATIONS
We utilize the phase-field model to simulate the microstructural evolution of the FMNS system by solving the Cahn–Hilliard [25] and Allen-Cahn (Ginzburg-Landau) equations [26] to simulate the microstructural evolution. We will denote the composition (i = 1, 2, 3, and 4 for Fe, Mn, Ni, and Si, respectively) in phase θ using c i θ
where [27],
where np represents the number of θ precipitate.
The local free energy density G(c i θ
or
where the units of all free energy densities (expressed by g in this study) in this study are joules per mole: the unit of total free energy of the system G is J. We assumed that density ρ is 7.9 g cm3 and the atomic molar mass M is 56.
(4)
And
with c 1 α = 1 . 0 - c 2 α - c 3 α - c 4 α
where y 3 I I y 4 I I
We implemented the forward Euler scheme to solve Eq. 9 and 10. To determine Di in Eq. 10 we adopted the relation in refs. [20,21].
We use the relation D i θ T = D i θ 0 e x p - Q i θ R T
In principle, one has to know the diffusivity data of each element of each precipitated phase to perform phase-field modeling. However, the diffusivity data of T3, T6, and T7 phases are not available. Since a precipitate larger than 17 nm has the FCC crystal structure [30], we adopted the diffusion data of each element in the Austenite (FCC) phase for all precipitated phases.
We could not find diffusivity data for Si in the γ(fcc) phase; therefore, we calculate it from a kinetic simulation. We performed a DICTRA simulation using Thermo-Calc 2017a software with the TCFE8 and MOBFE3 databases. At T = 550K, in the Fe–Si binary system (γ(fcc) phase) the Si mole fraction is always 0.03 at x = 0; the value is 0.005 initially, except for the point of x = 0 in Fig. 1. From the diffusion length depending on the simulat ion time, we calculated the diffusion coefficient of Si in the γ(fcc) phase.
In our simulations, we use the energy normalized by RT, where R = 8.3144598 J/mol·K and the temperature T = 550 K. The diffusivity values are normalized by D 2 α
To determine ωθ in Eq. 12, the interfacial energies between the matrix and precipitate phases are needed. So far, we have not found any literature that has evaluated the interfacial energies relevant for our study; therefore, we measure the interfacial energy using the extended Becker’s model function [31]. To perform the interfacial energy estimation, we used TC-PRISMA software (included in Thermo-Calc 2016a) with the TCAL3 and MOBAL3 databases. However, evaluating the interfacial energy for the whole Si fraction in the T6 phase using TC-PRISMA is not possible. We find that the Si fraction in the T6 phase is generally smaller than 0.3; therefore, we assume that the interfacial energy is given by Eq. 13. 0.2496 J/m2 [22]. For the T3 and T7 stoichiometric compounds, we also evaluate the interfacial energies using TC-PRISMA as 0.3311 and 0.1791 J/m2. The interfacial energy is given by Eq. 13 when the gradient coefficient is ε2.
Since we used the normalized variables in this study, we evaluated the normalized interfacial energy.
The unit of ε is J⁄m2⋅mol. We chose ε*,T6 = 1.0 as a non-dimensional parameter which corresponds to the interfacial energy 0.26 J/m2 which is quite comparable to the value 0.2496 J/m2 which we obtained from the TC-Prisma calculation. We also determined ε*,T3 = 1.33 and ε*,T7 = 0.72 using a consistent way.
4. SIMULATION DETAILS
We performed the simulations with a simulation cell size of 8 × 8 nm2. The initial precipitate radius was 1.5 nm. The discretized time step was selected as given in Table 2.
We chose Δt after convergence and accuracy testing. The simulations obtained results in 1 to 12 h, depending on the Δt of the system. For the matrix, the initial compositions in the α phase were c 2 α c 3 α c α c 3 T 6 = 0 . 459234067 c 4 T 6 = 0 . 2074326
For the T3 and T7 cases, the compositions of the precipitates were fixed and the initial compositions of the α phase were assumed to be equal to those of the T6 case. For the case with a single precipitate, we placed a circular precipitate with an initial radius r = 1.5 nm at the center of the simulation cell. For multiple precipitate phases, we placed two precipitates with initial radii r = 1.5 nm at two symmetrical positions with the distances L = 3.25, 3.50, and 3.75 nm relative to the center of the system.
5. RESULTS
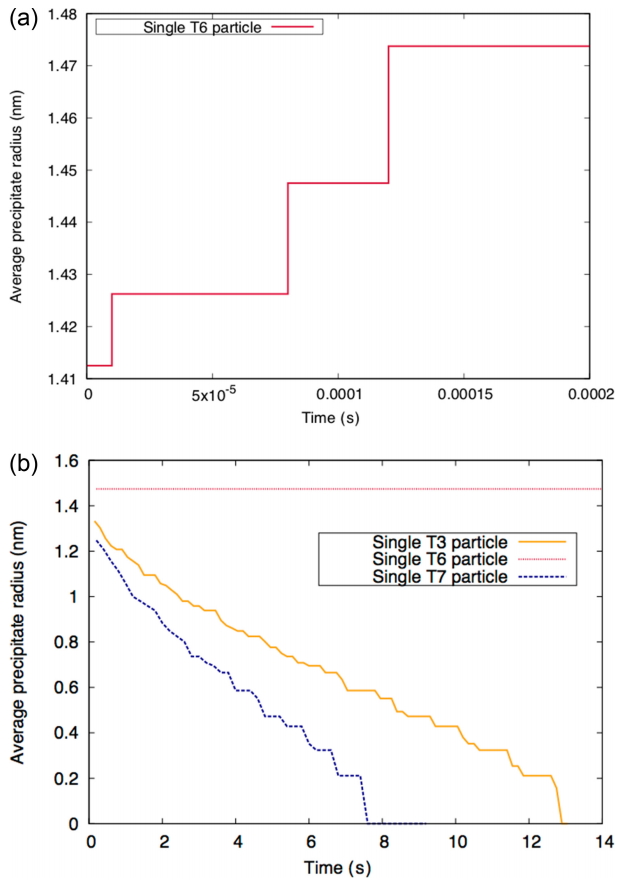
We performed the simulation to examine the stability of a single precipitate as shown in Fig. 2. In Fig. 3(a), the T6 precipitate undergoes minor accommodation at a very early stage. After 0.0002 s, the precipitate radii remains the same as it was during the modeling in Fig. 3(b). This means that the T6 precipitate is thermodynamically stable. Because the T3 and T7 phases are unstable, these precipitates shrink with time; the T7 precipitate radius decreases more rapidly than that of the T3 precipitate. We performed CALPHAD modeling using Thermocalc software and the UW1 database [16] with c 2 α c 3 α c 4 α
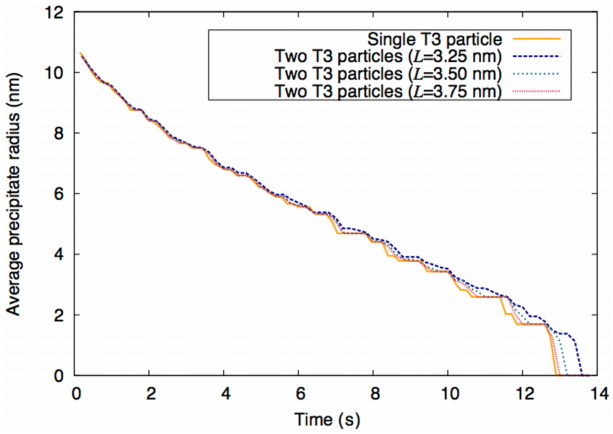
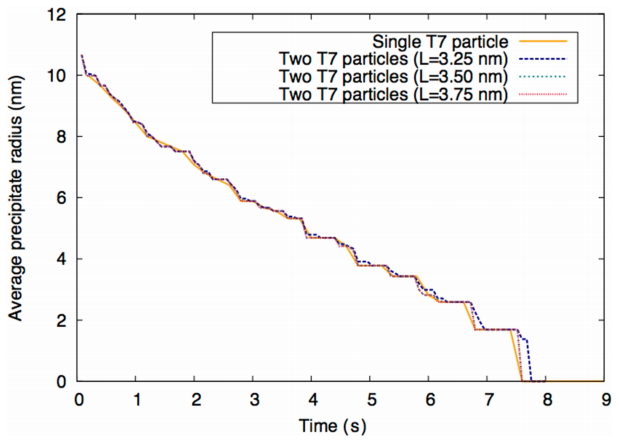
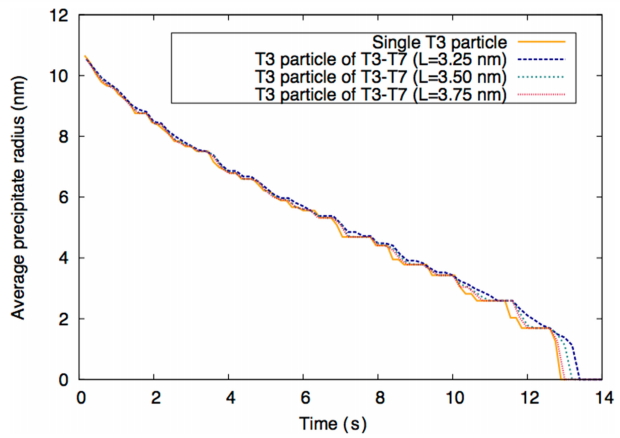
Therefore, the T3 particles remain longer for two interacting T3 particles compared to a single T3 particle. In addition, when the distance between particles L is 3.75 nm, the particle size evolution curve shown in Fig. 5 approaches convergence with that curve of the single T3 particle. Consistent results are observed for the T7 precipitate in Fig. 6.
6. CONCLUSIONS
We developed a multi-scale modeling framework to simulate the microstructural evolution of MNS precipitates in the quaternary FMNS system representing RPV steel. The UW1 quaternary database was implemented and the DICTRA and TC-PRISMA packages were used to evaluate the mobility of solutes and the interfacial energies between the matrix phase and precipitate phases. The results of the single-precipitate simulations were consistent with the thermodynamic model. We also evaluated the interactions between precipitates and found that the precipitates survived longer when two particles were present, whether of the same or different phases, compared to the particle lifetime in the single-precipitate case.