Porous MnO2/ carbon Hybrid Material with Improved Electrochemical Performance
Article information
Abstract
In this work, MnO2 nanoparticles were embedded in a carbon matrix as a porous composite, fabricated using a simple chemical route followed by low-temperature annealing, with activated carbon (AC) as the carbon source in the composite preparation. The porous MnO2/carbon structures contained some selective nanoparticles coated with carbon. The structural feature was identified by transmission electron microscopy (TEM). The surface area and pore size distribution of the materials were investigated by N2 adsorption/desorption isotherms, and demonstrated a high surface area of about 80 m2 g-1. AC is a readily available carbon source that can easily form a composite with MnO2 nanoparticles, forming a distinctive porous morphology. When employed as an anode material for lithium-ion batteries (LIB), the composite electrode demonstrated high specific capacities with an initial discharge capacity of 2500 mAh g-1 and maintained about 1391 mAh g-1 after fifty cycles. It also demonstrated excellent high rate performance, delivering more than 500 mAh g-1 of specific capacity at 3000 mA g-1, which is a higher capacity than a conventional graphite anode. Overall, the MnO2/ carbon composite electrode delivered superior anode performance, which was attributed to the improved surface area of the carbon hybridized MnO2 nanoparticles. The porous composite has benefits for lithium storage performance.
1. Introduction
Recently, researchers have conducted studies to develop a wide variety of nanoparticles and carbon composite electrode materials, to enhance the performance and capacity of lithium- ion secondary batteries (LIBs), their life cycle, and improve their rate performance. Metal composites, metal alloys and metal oxide carbon nanohybrid materials, with porous surface features, have attracted considerable attention for electrochemical studies and as battery electrodes because of their high particular surface area, good infusion, and electrolyte permeability [1-8]. These properties improve their specific capacities and rate performances, making them competitive [1-5]. MnO2 is the 12th most abundant element in earth’s crust. Its easy availability, low raw material cost and non-toxic nature makes MnO2 an attractive material compared to other transition metal oxides [10]. The different crystallographic morphologies of MnO2 including α, β, γ, and δ-types have been extensively used in various sectors, including catalysis, as electrode materials in batteries, in water treatment, and sensors [11-16]. Among these polymorphs, α-MnO2 has a distinctive [2 × 2] tunnel structure, while λ-MnO2 has a 3D spinel structure, and have received considerable attention as anode electrode materials in energy storage applications. λ and α polymorphs of MnO2 have been investigated as LIB anodes in previous reports [9,17]. Their noted advantages, such as their good electron transfer and lithium-ion diffusion of λ-MnO2, have been studied as an efficient electrode material for LIBs and supercapacitors [17-19]. However, the intrinsically poor electrical conductivity of MnO2 hampers the electronic pathway, and consequently results in capacity fading under prolonged cycles. This is a significant drawback of the MnO2 electrode [9,17,20]. To overcome these issues, environmentally benign and high-conducting carbon materials have been widely used as a composite with MnO2 to improve its electrochemical performance [20-23]. In this light, porous carbon materials such as activated carbon, graphene and carbon nanotubes etc. are usually studied due to their high electrical conductivity and large specific surface area.
Among these, activated carbon (AC) can be a first-rate carbon template for nanocomposite materials as LIB anodes, due to its easy availability, large surface area with adjustable porosity, low cost, and chemical stability [24]. Previously, limited results were reported for MnO2/ activated carbon hybrid nanostructures prepared by different chemical approaches, and studied as electrode materials for supercapacitors [25-28]. Choi et al., prepared a MnO2/ activated carbon composite by dispersing MnO2 and carbon particles separately in deionized water, followed by hydrothermal treatment and annealing at 400 °C [26]. Deng et al., reported AC derived from human hair and a KMnO4 mixture, which was stirred at 60 °C for 24 h and later vacuum filtered [27]. In another report from Wang et al., a functionalized AC powder mixed Mn(Ac)2 and KMnO4 separately and subjected them to hydrothermal treatment to attain AC/MnO2 composites [28]. These reported methods involved complex hydrothermal synthetic processes and long reaction processing times to achieve nanocomposites. Also, it should be noted that these MnO2/ activated carbon composites were mainly studied as electrodes for supercapacitors, with barely any studies on MnO2/ AC as anodes for LIBs. Combining MnO2 and AC may provide exceptional electrical conductivity and electrochemical stability to the AC, with the high specific capacity of MnO2 (the oretical capacity for an Li-ion battery anode is 1223 mAh g-1).
The hybrid electrode electrochemical properties mainly depend on the morphology of the MnO2 composite, the nature of the carbon source. Controlling the morphology is a challenging task that depends on the synthesis method employed for carbonization. As noted, the synthesis methods to prepare MnO2/AC composite involve multi-step processes, and also raise issues related to choosing a suitable carbon precursor, pretreatment of the carbon precursors, and avoid high heating temperatures that can instead cause secondary metal oxide phase formation. The high active surface area and porosity of the nanosized MnO2 arrays play an essential role in determining the resultant composite electrochemical properties and cycling stability. We employed high surface area activated carbon as the carbon source in the current work to meet these requirements.
We prepared a nanocomposite material of MnO2/ porous carbon by employing a simple wet impregnation method, followed by low-temperature annealing, in which MnO2 nanoparticles are embedded within a carbon matrix with a porous surface morphology. The prepared nanocomposite material was studied as a negative electrode for LIB.
2. Experimental Procedures
2.1 Preparation of the MnO2/ C composite
In a typical synthesis approach, 100 mg of AC powder (PCT-AC-07, BET: 2000-2,300 m2 g-1) was mixed with Mn (NO3)2·6H2O (Aldrich, > 99.9%), and an appropriate amount of isopropyl alcohol was added to form a paste (ACs/ Mn (NO3)2·6H2O = 1:2), which was then mixed using a mini ball mill (Fritsch, pulverisette 23) for approximately 20 m. at a speed of 30 oscillations/s. to form a homogeneous mixture. The resulting mixture was collected and heat-treated in a tube furnace at 400 °C for 1 h under an Ar atmosphere. The obtained product was labeled C-MnO2. The formation of the MnO2/ carbon hybrid structures is shown in scheme (1).
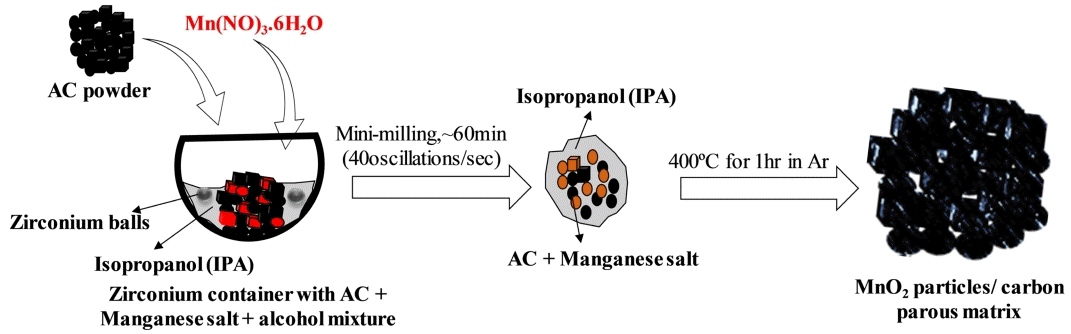
(a) Schematic of the porous C-MnO2 composite synthesis process.
As shown in scheme 1, Mn(NO3)2·6H2O is homogeneously soluble in isopropanol at ambient temperature and forms a uniform mixture with AC powder after 60 min of mini milling. During the process of milling, there is the possibility of mild nucleation of Mn2+ oxidation on AC solid powder, as MnOOH/ MnO2 particles in the alcohol solution. After annealing, the resultant product consisted of MnO2 particles combined with a porous carbon matrix.
2.2 Characterization
The surface morphology and microstructures of the C-MnO2 were examined by SEM (scanning electron microscopy, JSM7000F, JEOL, Japan), HR-TEM (high-resolution transmission electron microscopy, JEM-2100F), and energy-dispersive X-ray (EDX) spectroscopy. The crystal structure of the prepared materials was investigated by powder X-ray diffraction (XRD) using a Rigaku D/MAX- 2200 Ultima diffractometer with Cu-Kα radiation (λ = 1.54056 Å) with in the range of 10°< 2θ < 80° operating at 30 kV and 40 mA. N2 adsorption-desorption isotherms were measured using a Micromeritics ASAP ZOZO instrument at 77 K, and the Brunauer calculated surface areas–Emmett– Teller (BET) method.
2.3 Electrochemical measurements
The MnO2/ carbon composite electrode performance was measured with a CR 2032 coin cell. The electrode composition was comprised of 85% active material, 5% conductive agent (Ketjen Black) with 10% PAI (polyamide-imide) binder dissolved in N-methyl pyrrolidone (NMP) solution. Then the slurry was uniformly coated on Cu foil using the doctor-blade method. The coated foil was dried at 80 °C for 3 h to evaporate the solvent and then vacuum dried for 3 h at 200 °C to activate the binder. The coin-type half-cells were assembled in Ar filled glove box, using 1 M LiPF6 in EC/ DMC 1:1 as an electrolyte with Celgard 2300 membrane as the separator and metallic lithium as a counter electrode. The charge/discharge experiments were studied at different applied current densities of 100-3000 mA g-1 within the voltage range of 0.01-3.0 V vs. Li+/ Li under ambient conditions. Cyclic voltammetry (CV) was performed under the same voltage range at a scan rate of 0.1 mV s-1.
3. Results and Discussion
The prepared material was investigated by XRD; all diffraction peaks correlated well with the formation of γ-MnO2 (ICDD-JCPDS No. 14-0644), representing good crystallinity in the prepared product (as shown in Fig 2(a)). The typical carbon (002) peak noted around 20-25° of 2θ possibly resembles the (120) peak of γ-MnO2. Fig 2(b) shows a panoramic TEM image of the as-synthesized C-MnO 2 sample. The sample is comprised of well-formed aggregated MnO2 nanoparticles of about 10-50 nm in size; those are connected by randomly distributed void spaces of about <10 nm between the aggregated particles. Notably, some particles are coated with a thin-layer of carbon as shown in the inset of Fig 2(b), and we expect some portion of the AC powder and isopropanol fused onto the metal oxide particles during the annealing treatment, which results in a coated carbon layer. The nano-thin coated carbon layer (<10 nm, see inset of Fig 2(b)) on MnO2 particles can undoubtedly improve the physical and electrochemical properties of the host nanoparticles, as was witnessed in previous works [5,21,31].
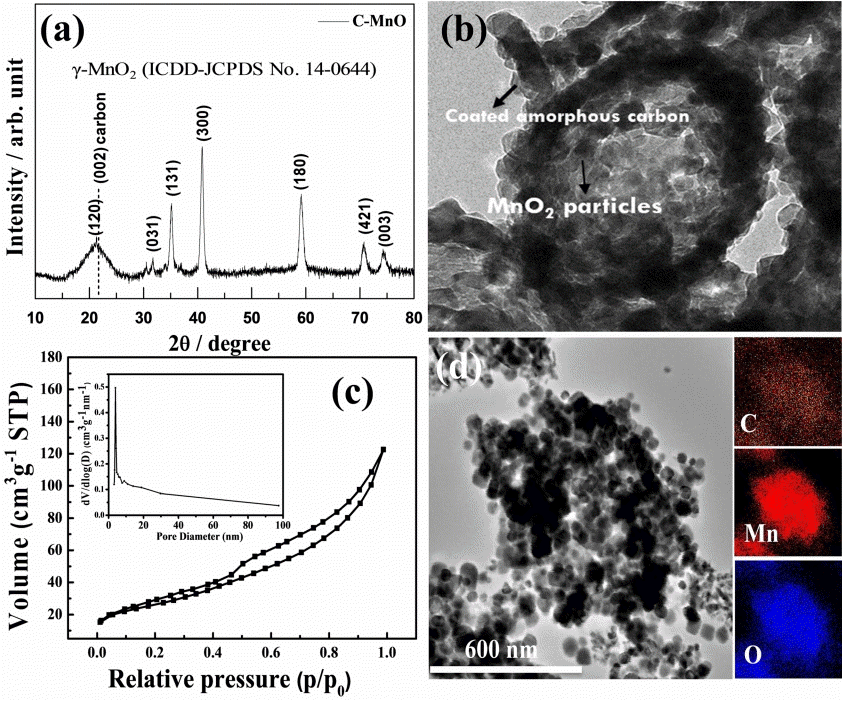
(a) XRD, (b) TEM with inset HR-TEM, (c) N2 adsorption-desorption isotherms, and the inset is BJH pore size distribution curve (d) HR-TEM and its corresponding EDX spectrum of the MnO2/C composite powder.
The TEM image clearly shows the particles’ porous nature, interconnected along with the carbon matrix. The porosity of the C-MnO2 sample was analyzed by N2 adsorption/desorption isotherms. Fig 2(c) shows a typical type IV isotherm with H3 hysteresis loop, which appears to be cavitation induced evaporation. This seems to be common in many micro/ mesoporous materials because of the gap resulting from the formation of aggregations of nanoparticle/carbon particles [32]. This is consistent with the observation in the SEM and TEM images.
Further, the inset in Fig 2(c) shows the pore size distribution analysis, confirming that the porous C-MnO2 formed. The majority of pore sizes was found to be 3.89 nm alongside with the minor probability pore size of which estimated from the other curves are about 7.8 and 13.8 nm. The specific surface area and pore volume were 90.49 m2 g-1 and 0.175 cm3g-1, respectively. The elemental mapping of the C-MnO2 was estimated by TEM-EDX analysis; the inset images in Fig 2(d) show the presence of Mn, O, and C within the C-MnO2 sample, where MnO2 nanoparticles of less than 50 nm size are interconnected with each other alongside the carbon matrix. The mapping result indicates the distribution of carbon in and around the MnO2 particles embedded in a porous carbon matrix. The SEM image in Fig 3(a) shows a group of well-grown MnO2-carbon porous structures with some particular forms with diameters of around 200-500 nm (marked with a colored circle in Fig 3(a)). Fig 3(b) is the SEM elemental mapping of C-MnO2, which displays the presence of the yellow color carbon dots all over the measured sample, along with Mn and O dots, indicating the equal distribution of carbon with MnO2. Also, about 26% of the carbon content appeared in the C-MnO2 sample’s TGA graph (as shown in Fig 3(c)).
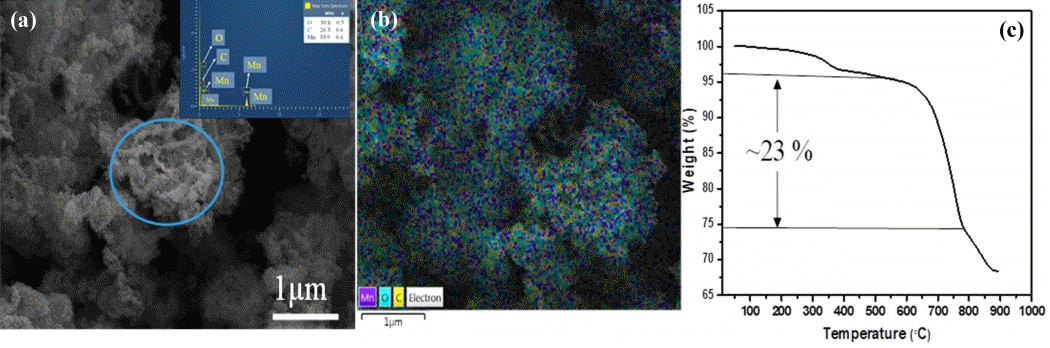
(a) SEM and inset EDS and (b) elemental mapping (c) TGA graph of the C- MnO sample.
Fig 4(a) illustrates the cycling performance when initially applied as an anode electrode for LIB, with their corresponding coulombic efficiencies (CEs) of C-MnO2 electrodes at 100 mA g-1 of applied current density within the voltage range of 0.01-3 V (vs. Li/Li+) for 50 cycles. The electrode exhibited a high initial discharge and charge capacities of 2500 and 1654 mAh g-1, respectively, with 66% initial CE. Notably, the initial discharge capacity was much higher than the theoretical specific capacity of MnO2 (i.e., 1232 mAh g-1). This considerably large extra specific capacity of the MnO2 electrode is attributed to the presence of a gel-like SEI film organic and inorganic compounds (LiF, Li2CO3, and ROCO2Li) formed on the fresh electrode [33].

(a) Cycling stability of the C-MnO2 electrode tested within the voltage range of 0.01-3 V at 100 mAh g-1 (b) and the respective 1st, 2nd and 25th cycling profiles, (c) CV curves of the C-MnO2 electrode at a scan rate of 0.1 mV s-1 between 0.01 and 3 V, (d) Rate capability of C-MnO2 electrode at different current densities and inset shows the voltage profiles cycled at 2000 and 3000 mA g-1.
The discharge-charge capacities decreased to 1738 and 1658 mA g-1 in the second cycle (CE was 95%); after the 5th cycle, the capacities stabilized at 1244 and 1213 mAh g-1 (CE was 97%) until the 20th cycle (CE close to 97%). A gradual increase in specific capacity was observed for the rest of the cycles, and after 50 cycles, it finally stabilized at about 1391 and 1360 mAh g-1, respectively. The electrode presented near unity CE after a few cycles, indicating the excellent reversibility of the electrode reactions. The corresponding C-MnO2 voltage profiles of the 1st, 2nd and 25th are displayed in Fig 4(b). The two shoulders near ~1.4 V and ~2.5 V in the charging process resemble the Mn/MnO and MnO/Mn3O4 redox potentials; a flat voltage plateau was located at about 0.5 V, which is the typical voltage charge-discharge profile trend noticed for MnO2 electrodes [17].
Fig 4(c) shows the cyclic voltammograms (CV) of the C-MnO2 for ten consecutive cycles within the potential range of 0.01-3V. Characteristic features in the CV profiles include significant peaks at 0.28 V in the cathodic scan, accredited to the formation of a solid electrolyte interphase (SEI) layer. The bulky cathodic peak at ~0.008 V in the first cycle corresponds to the reduction of Mn. In consecutive cycles, the initial cathodic peak shifts to a positive potential of ~0.27 V, which is ascribed to the structural reconstruction resulting from the formation of Li2O and metallic manganese through the conversion reaction given in Equation (1) [17, 26].
The broader CVs with little intensified redox peaks indicate the improved electrochemical accessibility of the electrolyte, possibly due to increasing electrical conductivity, induced by the carbon coating on the MnO2 particles. In the anodic direction, the peaks were produced at 1.5 V due to the reversible oxidation of metallic Mn, by following MnO2 redox processes, as mentioned in a previous report [9].
The rate performance of the C-MnO2 electrode is shown in Fig 4(d); the C-MnO2 electrode exhibited discharge/ charge capacities of 1998/1281, 1046/1035.6, 864.16/862.9, 740/ 735.20, 701.8/700.2, 649.3/646.87, 599/598.32 and, 959/ 934.6 mAh g-1 at applied current densities of 100, 200, 300, 500, 1000, 2000, 3000 and 200 mAh g-1, respectively. Interestingly, at high current densities of 1000, 2000 and, 3000 mAg-1, C-MnO2 delivered good capacities, higher than 590 mAh g-1, which is about two-fold higher than a typical graphite anode (372 mAh g-1). The noted irreversible capacities of C-MnO2 for the first cycles measured at 2000 and 3000 mAg-1 were about 34.05 and 2.51 mAh g-1 and for the following cycles its CE values were close to unity. This is a typical perceptible performance for anodes, particularly during swift cycling to higher current densities.
Further again at 200 mAh g-1 the electrode retained a reversible capacity of about 934 mAh g-1, demonstrating the excellent rate performance of this material. The respective cycle profiles at 2000 and 3000 mAg-1 are shown in the inset of Fig 4(d), which displays similar voltage curves, like those noted at low current rates (see Fig 4(b)) with no further new peaks. This explains the stability of the electrode at high current rates.
The obtained rate performance of our C-MnO2 electrode was better than the reported work on MnO2 anode electrodes [9,17,33-36]. Compared to other MnO2-based anodes [36-41], our C-MnO2 electrode delivered excellent rate capabilities and much more stable cyclability (Fig 5). The obtained results showed that our electrode exhibited excellent cycle stability and remarkable rate performance. This confirms that the C-MnO2 composite with MnO2 nanoparticles are individually connected as a hierarchical framework with a high surface area porous carbon network. This kind of structure offers a large electrolyte/ electrode contact area. Subsequently, this can facilitate Li-ion passage through the C-MnO2 electrode. The conductive carbon network improves electron transfer to the maximum extent, which benefits the rate performance. The interconnected MnO2 nanoparticles/ carbon framework arrangement provides electrodes better stability against high current densities and maintains better rate performance and good cycle life.
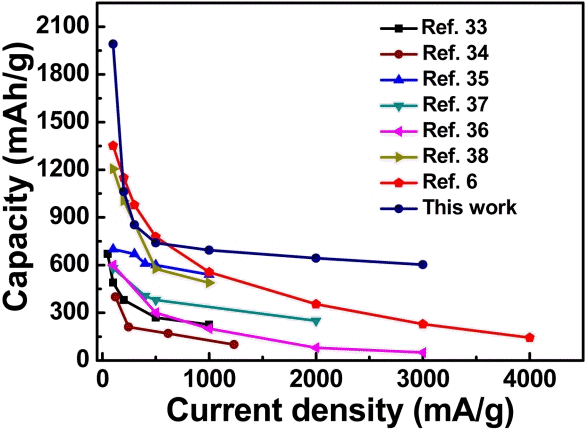
Evaluation of the rate performances of the C-MnO2 electrode and the other reported MnO2-based anodes for Li-ion batteries.
4. Conclusion
In summary, we successfully synthesized MnO2 nanoparticles in an interconnected framework embedded in a porous carbon matrix, using a simple chemical method along with mini-milling process and low-temperature annealing treatment, for use as an anode material in LIBs. The electrochemical performance revealed that the given porous MnO2 /carbon composite displayed an improved specific capacity with decent cycling stability as an anode material. It was presumed that the superior electrochemical performance resulted from the combination of MnO2 nano structural features, along with a porous carbon framework which enhanced the maximum lithium-ion passage, improved the charge-transport process and electrode stability, leading to excellent cyclability and superior rate capability. The porous MnO2 /carbon composite shows great potential for use as high-efficiency anodes in LIBs, and can be extended as an anode material for other energy storage devices like lithiumair batteries and super capacitors. In addition, the present approach can be easily adapted to prepare porous other metal oxides and carbon composites with tremendous performance.
Acknowledgements
This work was supported by the National Research Foundation of Korea (NRF) grant funded by the Ministry of Education (NRF-2018R1D1A1B07044026).
Notes
The authors declare no conflict of interest.