1. ņä£ ļĪĀ
ņŚÉļäłņ¦Ć ņé¼ņÜ® ĻĖēņ”ØĻ│╝ ĒÖöņäØņŚ░ļŻīņØś Ļ│╝ļÅäĒĢ£ ņé¼ņÜ®ņ£╝ļĪ£ ņØĖĒĢ£ ņ¦ĆĻĄ¼ ņś©ļé£ĒÖöņÖĆ ĒÖśĻ▓ĮņśżņŚ╝ņØś ņśüĒ¢źņ£╝ļĪ£ ĒÖśĻ▓Įņ╣£ĒÖöņĀü ņŚÉļäłņ¦Ć ņāØņé░ ļ░®ņŗØņØĖ ņŚ░ļŻīņĀäņ¦ĆņŚÉ ļīĆĒĢ£ Ļ┤Ćņŗ¼ņØ┤ ņ”ØĻ░ĆĒĢśĻ│Ā ņ׳ļŗż. ĒŖ╣Ē׳, Ļ│ĀļČäņ×É ņĀäĒĢ┤ņ¦ł ņŚ░ļŻīņĀäņ¦Ć(proton exchange membrane fuel cell, PEMFC)ļŖö ļ╣äĻĄÉņĀü ņĀĆņś©ņŚÉņä£ ņÜ┤ņĀä Ļ░ĆļŖźĒĢśļ®░ ņŚÉļäłņ¦Ć ļ│ĆĒÖś ĒÜ©ņ£©ņØ┤ ļåÆĻ│Ā, ņ£ĀĒĢ┤ Ļ░ĆņŖżļź╝ ļ░░ņČ£ĒĢśņ¦Ć ņĢŖļŖöļŗż. ņØ┤ļĪ£ ņØĖĒĢ┤ PEMFCļŖö ĒÖöņäØņŚ░ļŻīļź╝ ļīĆņ▓┤ĒĢĀ ņ▓ŁņĀĢ ņŚÉļäłņ¦Ć ļ░£ņĀäņ£╝ļĪ£ ņŻ╝ļ¬® ļ░øĻ│Ā ņ׳ļŗż [1,2].
ļ░▒ĻĖł(Pt)ņØĆ PEMFCņØś ņłśņåīņé░ĒÖöļ░śņØæ ļ░Å ņé░ņåīĒÖśņøÉļ░śņØæņŚÉ ļ¦żņÜ░ ļø░ņ¢┤ļé£ ņ┤ēļ¦ż ĒÖ£ņä▒ņØä ļ│┤ņØĖļŗż [3,4]. ļśÉĒĢ£ ņĀäĻĖ░ņĀü ņĀäļÅäņä▒ņØ┤ ļø░ņ¢┤ļéśĻ│Ā ĒÖöĒĢÖņĀüņ£╝ļĪ£ ņĢłņĀĢĒĢśņŚ¼ ņĀäĻĖ░ĒÖöĒĢÖ ņ┤ēļ¦żļĪ£ ļ¦żņÜ░ ņżæņÜöĒĢśļŗż. ņé░ņåīĒÖśņøÉļ░śņØæ(oxygen reduction reaction, ORR)ņØś ņåŹļÅäļŖö ņłśņåīņé░ĒÖöļ░śņØæ(hydrogen oxidation reaction, HOR)ņØś ņåŹļÅäņŚÉ ļ╣äĒĢ┤ ļ¦żņÜ░ ļŖÉļĀż PEMFCņØś ņĀäņ▓┤ ņä▒ļŖźņØä Ļ▓░ņĀĢĒĢ£ļŗż [5-8]. ĻĘĖļ¤¼ļéś PtļŖö ņ¦ĆĻĄ¼ ņāüņŚÉ ņåīļ¤ē ņĪ┤ņ×¼ĒĢśļŖö Ļ│ĀĻ░ĆņØś ĻĘĆĻĖłņåŹņØ┤ļŗż. ļśÉĒĢ£ ļé«ņØĆ pHņÖĆ Ļ│ĀņĀäņĢĢņŚÉņä£ ņÜ┤ņĀäļÉśļŖö PEMFCņØś ĒŖ╣ņä▒ņāü ĻĖłņåŹņØś ņĢłņĀĢņä▒ņŚÉ Ļ▓░ĒĢ©ņØ┤ ņāØĻĖ░ļ®░, ņØ┤ļĪ£ ņØĖĒĢ┤ ņ×ģņ×É Ēü¼ĻĖ░ņØś ņ”ØĻ░Ć [9], ņ¦Ćņ¦Ćņ▓┤ ĒāäņåīņØś ļČĆņŗØ [10], Pt ņÜ®ņČ£ [11] ļō▒ņØś ļ¼ĖņĀ£Ļ░Ć ļ░£ņāØĒĢ£ļŗż.
Pt ņ┤ēļ¦żņØś ļŗ©ņĀÉņØä ĒĢ┤Ļ▓░ĒĢśĻĖ░ ņ£äĒĢ£ ļīĆņĢł ņżæ ĒĢśļéśļŖö Ptļź╝ ņĀüņĀłĒĢ£ ņ¢æņØś ņĀäņØ┤ĻĖłņåŹ(M)Ļ│╝ ĒĢ®ĻĖłĒĢśļŖö Ļ▓āņØ┤ļŗż. ņ¦Ćļé£ ņłśņŗŁļģäĻ░ä ļ¦ÄņØĆ ņŚ░ĻĄ¼ņ×ÉļōżņØ┤ Pt-MņØä ĒĢ®ņä▒ĒĢśņŚ¼ ņ┤ēļ¦żņØś Ļ░ĆĻ▓®ņØä ļé«ņČöĻ│Ā ORRņŚÉ ļīĆĒĢ£ ĒÖ£ņä▒Ļ│╝ ļé┤ĻĄ¼ņä▒ņØä Ē¢źņāüņŗ£ĒéżĻĖ░ ņ£äĒĢ£ ļŗżņ¢æĒĢ£ ņŗ£ļÅäļź╝ ĒĢśĻ│Ā ņ׳ļŗż. Pt-Ni [12], Pt-Co [13], Pt-Fe [14] ļō▒ņØś 3d ņĀäņØ┤ĻĖłņåŹ ĒĢ®ĻĖłņØ┤ ĻĘĖ ļīĆĒæ£ņĀüņØĖ ņśłņØ┤ļŗż.
ņØ┤ļ¤¼ĒĢ£ Pt-M ņ┤ēļ¦żņØś ĒÖ£ņä▒ņØĆ active area, Ēæ£ļ®┤ņØś ņøÉņ×ÉĻĄ¼ņĪ░, Ēü¼ĻĖ░, ĒśĢĒā£ ļō▒ņŚÉ ņśüĒ¢źņØä ļ░øļŖö Ļ▓āņ£╝ļĪ£ ņĢīļĀżņĀĖņ׳ļŗż [15]. ņØ┤ļĪ£ ņØĖĒĢ┤, Pt-Pt ņøÉņ×ÉĻ░ä Ļ▒░ļ”¼Ļ░Ć Ļ░ÉņåīĒĢśĻ│Ā, d-band centerņØś ņØ┤ļÅÖņ£╝ļĪ£ Pt-O ļ░Å O-O Ļ▓░ĒĢ®ņØä ņĢĮĒÖöņŗ£ņ╝£ ORR ĒÖ£ņä▒ņØä Ļ░£ņäĀĒĢĀ ņłś ņ׳ļŗż [2,3,16,17]. ļśÉĒĢ£, PtĻ│╝ ņĀäņØ┤ĻĖłņåŹņØś ņāüĒśĖņ×æņÜ®ņ£╝ļĪ£ ņŗ£ļäłņ¦Ć ĒÜ©Ļ│╝ļź╝ ņØ╝ņ£╝ĒéżĻ│Ā bi-functional ļ®öņ╗żļŗłņ”śņ£╝ļĪ£ ņØĖĒĢ┤ ņł£ņłśĒĢ£ Pt ņ┤ēļ¦żļ│┤ļŗż ņÜ░ņłśĒĢ£ ņĀäĻĖ░ĒÖöĒĢÖ ņ┤ēļ¦ż ĒÖ£ņä▒ņØä ļ│┤ņØĖļŗż [18].
Pt-MņØś ņä▒ļŖźņØĆ ņ┤ēļ¦ż ņ¦Ćņ¦Ćņ▓┤ņØś ņóģļźśņŚÉ ņśüĒ¢źņØä ļ░øļŖöļŗż. ļåÆņØĆ Ēæ£ļ®┤ņĀü, ĒÖöĒĢÖņĀü ņĢłņĀĢņä▒Ļ│╝ ņÜ░ņłśĒĢ£ ņĀäļÅäņä▒ņØä Ļ░¢ļŖö Ēāäņåī ņ¦Ćņ¦Ćņ▓┤ļź╝ ņŻ╝ļĪ£ ņé¼ņÜ®ĒĢ£ļŗż [2,19-21]. ĒĢśņ¦Ćļ¦ī, PEMFC ņÜ┤ņĀä ņŗ£ņ×æĻ│╝ ļ®łņČż ņŗ£ Ēāäņåī ņ¦Ćņ¦Ćņ▓┤Ļ░Ć ļČĆņŗØļÉĀ ņłś ņ׳ļŗż. Ēāäņåī ņ¦Ćņ¦Ćņ▓┤Ļ░Ć ļČĆņŗØļÉ£ ņ┤ēļ¦żļŖö Pt ļśÉļŖö Pt-M ņ┤ēļ¦ż ņ×ģņ×ÉĻ░Ć ļČłņĢłņĀĢĒĢ£ ņāüĒā£ļĪ£ ļéśļēśņ¢┤ ļ¢©ņ¢┤ņĀĖ PEMFC ņä▒ļŖźņØä ņĀĆĒĢśņŗ£ĒéżļŖö Ļ▓░Ļ│╝ļź╝ Ļ░ĆņĀĖņś©ļŗż [22]. ņØ┤ļ¤¼ĒĢ£ ļ¼ĖņĀ£ļź╝ ĒĢ┤Ļ▓░ĒĢśĻ│Āņ×É ņ¦łņåī ļō▒ņØś ņøÉņåīļź╝ ļÅäĒĢæĒĢ£ Ēāäņåī ņ¦Ćņ¦Ćņ▓┤ [23], ĻĘĖļלĒĢĆ [24], ņ╣┤ļ│Ė ļéśļģĖ ĒŖ£ļĖī [25] ļō▒ ļŗżņ¢æĒĢ£ ņ¦Ćņ¦Ćņ▓┤ Ļ┤ĆļĀ© ņŚ░ĻĄ¼Ļ░Ć ņ¦äĒ¢ēļÉśĻ│Ā ņ׳ļŗż.
ņŚ┤ņ▓śļ”¼ļŖö ņ┤ēļ¦żņØś ņ×ģņ×É Ēü¼ĻĖ░, ļČäņé░ļÅä, ĒĢ®ĻĖłļÉśļŖö ņĀĢļÅä ļō▒ņØä ņĪ░ņĀłĒĢśĻ│Ā ņ¦Ćņ¦Ćņ▓┤ Ēæ£ļ®┤Ļ│╝ ĻĄ¼ņĪ░ ĒŖ╣ņä▒ņŚÉ ņśüĒ¢źņØä ļ»Ėņ│É ņ┤ēļ¦żņØś ņĢłņĀĢņä▒ņØä Ļ░£ņäĀĒĢĀ ņłś ņ׳ļŗż [26]. Chaisubanan ĻĘĖļŻ╣ņŚÉ ļö░ļź┤ļ®┤, Pt/C ņ┤ēļ¦żņÖĆ ļ╣äĻĄÉĒĢśņŚ¼ Pt-M(M=Cr, Co, Pd) ņ┤ēļ¦żļŖö ņŚ┤ņ▓śļ”¼ Ēøä Pt-M Ļ▓░ņĀĢ ĻĄ¼ņĪ░Ļ░Ć ļ¦ÄņĢäņĪīĻ│Ā, Pt ņ×ģņ×ÉĻ░Ć ņØæņ¦æļÉśņ¢┤ ņĀäĻĖ░ ĒÖöĒĢÖ Ēæ£ļ®┤ņĀü(electrochemically active surface area, ECSA)ņØä Ļ░Éņåīņŗ£ņ╝░ļŗż [27]. ļśÉĒĢ£, ņŚ┤ņ▓śļ”¼ļŖö ņ┤ēļ¦żņØś ņĀäĻĖ░ ņĀäļÅäņä▒ņØä ņ”ØĻ░Ćņŗ£ņ╝░ņ£╝ļ®░ Ļ▓░Ļ│╝ņĀüņ£╝ļĪ£ Pt-M/C ņ┤ēļ¦żņØś ORR ĒÖ£ņä▒Ļ│╝ ņĢłņĀĢņä▒ Ļ░£ņäĀņŚÉ ņśüĒ¢źņØä ņŻ╝ņŚłļŗż.
ļ│Ė ļģ╝ļ¼ĖņŚÉņä£ļŖö ņĄ£ĻĘ╝ ņĢĮ 5ļģä ļé┤ņÖĖņŚÉ ļéśņś© Pt-M Ļ┤ĆļĀ© ļģ╝ļ¼ĖļōżņØä ņĀĢļ”¼ĒĢśņśĆļŗż. ņØ┤ļź╝ ĒåĄĒĢ┤ ļ░öņØ┤ļ®öĒāł ņ┤ēļ¦żņØś ņä▒ļŖźņŚÉ ņĀäņØ┤ĻĖłņåŹņØ┤ ļ»Ėņ╣śļŖö ņśüĒ¢źĻ│╝ ĒĢ®ĻĖłņØś ļ®öņ╣┤ļŗłņ”ś, Pt-MņŚÉ ņĀüņÜ®ļÉ£ ļŗżņ¢æĒĢ£ ņŗ£ļÅä ļō▒ņØä ņĢīņĢäļ│┤ņĢśļŗż. ļŗżņ¢æĒĢ£ ļ░öņØ┤ļ®öĒāł Pt-M ņ┤ēļ¦ż ņżæņŚÉņä£ļÅä Pt-Ni, Pt-Co, Pt-FeĻ░Ć Ļ░Ćņן ļ¦ÄņØ┤ ņŚ░ĻĄ¼ļÉśņŚłņ£╝ļ®░, PtņÖĆ MņØś ļ╣äņ£©, ņŚ┤ņ▓śļ”¼ ņĪ░Ļ▒┤, ņ¦Ćņ¦Ćņ▓┤ļź╝ ļ│ĆĻ▓ĮĒĢśņŚ¼ ņ┤ēļ¦ż ņä▒ļŖźņØä ļåÆņØ┤ļŖö ņŚ░ĻĄ¼Ļ░Ć ļ¦ÄņĢśļŗż.
2. ļ│Ė ļĪĀ
2.1 Pt-Ni ĒĢ®ĻĖłņ┤ēļ¦ż
Pt-Ni ņ┤ēļ¦ż ĒĢ®ņä▒ņŚÉļŖö ņØ╝ļ░śņĀüņ£╝ļĪ£ ņŚ╝ĒÖöļŗłņ╝ł [18]ņØä ņĀäĻĄ¼ņ▓┤ļĪ£ ņé¼ņÜ®ĒĢśļ®░, ĻĘĖ ņÖĖņŚÉļÅä ļŗłņ╝ł(II) ņĢäņäĖĒŗĖņĢäņäĖĒåĀļäżņØ┤ĒŖĖ [1] ļō▒ņØä Ni ņĀäĻĄ¼ņ▓┤ļĪ£ ņØ┤ņÜ®ĒĢśļŖö ņŚ░ĻĄ¼ļōżņØ┤ ļ│┤Ļ│ĀļÉśņŚłļŗż. Pt-Ni ņ┤ēļ¦żļŖö ļ®öĒāäņś¼ ņé░ĒÖö ļ░śņØæ(methanol oxidation reaction, MOR)Ļ│╝ ORR ļÅÖņ×æ ņĀäņ£äņŚÉņä£ņØś ņÜ®ņČ£ņØä Ļ░Éņåīņŗ£Ēéżļ®░, PtņØś Fermi energy ļĀłļ▓©ņØä Ļ░Éņåīņŗ£ņ╝£ Pt Ēæ£ļ®┤ņŚÉ COĻ░Ć ņēĮĻ▓ī ĒØĪņ░®ĒĢśņ¦Ć ļ¬╗ĒĢśĻ▓ī ĒĢśņŚ¼ ņ┤ēļ¦żņØś ņĢłņĀĢņä▒ņØä Ļ░£ņäĀĒĢśļŖö Ļ▓āņ£╝ļĪ£ ņĢīļĀżņĀĖ ņ׳ļŗż [28]. ĻĘĖļ¤¼ļéś, ņé░ņåī ņ╣£ĒÖöņä▒ ĻĖłņåŹņØ┤ Ļ│╝ĒĢśĻ▓ī ņĪ┤ņ×¼ĒĢĀ Ļ▓ĮņÜ░ PtņØś ĒÖ£ņä▒ ļČĆņ£äļź╝ Ļ░Éņåīņŗ£ņ╝£ ņ┤ēļ¦ż ņä▒ļŖźņŚÉ ņśüĒ¢źņØä ņżä ņłś ņ׳ļŗż. ļö░ļØ╝ņä£ ņé░ņåī ņ╣£ĒÖöņä▒ ĻĖłņåŹņØĆ Pt-M ņ┤ēļ¦żņŚÉ ņåīļ¤ē ļśÉļŖö ļ»Ėļ¤ē ņ▓©Ļ░ĆĒĢ┤ņĢ╝ĒĢ£ļŗż [29-31].
ļ¦ÄņØĆ ņŚ░ĻĄ¼ņ×ÉļōżņØ┤ Pt-NiņØś ņĄ£ņĀüņØś ĒĢ®ĻĖł ļ╣äņ£©ņØä ņ░ŠĻĖ░ ņ£äĒĢ£ ņŗżĒŚśņØä ĒĢśņśĆļŗż. PtxNi/C (x = 1, 2, 3, 5)ļź╝ ņĢīņ╣╝ļ”¼ ņĀäĒĢ┤ņ¦łņŚÉņä£ ņ┤ēļ¦ż ņä▒ļŖźņØä ņĖĪņĀĢĒĢ£ Ļ▓░Ļ│╝, ņĄ£ņĀüņØś ĒĢ®ĻĖł ļ╣äņ£©ņØĆ Pt:Ni=3:1ņØ┤ņŚłņ£╝ļ®░, Pt-Ni ĒĢ®ĻĖłņØ┤ ORR ļ░Å HER, HORņŚÉ ļīĆĒĢ£ ļ░śņØæ ņåŹļÅäļź╝ Ē¢źņāüņŗ£ĒéżļŖö Ļ▓āņØä ĒÖĢņØĖĒĢśņśĆļŗż [32]. ņäĖĻ░Ćņ¦Ć ļŗżļźĖ ņøÉņ×Éļ╣ä(Pt:Ni = 1:4, 1.4:1, 2.8:1)ļĪ£ ĒĢ®ņä▒ĒĢ£ Pt-Ni ņ┤ēļ¦żļź╝ ļŗżņżæļ▓Į Ēāäņåī ļéśļģĖĒŖ£ļĖī(multi-walled carbon nanotubes, MWCNTs)ņŚÉ ļŗ┤ņ¦ĆĒĢ£ ņŚ░ĻĄ¼ņŚÉņä£ļŖö, Pt:NiņØś ņøÉņ×Éļ╣äĻ░Ć 1:2.8ņØ╝ļĢī ņĄ£ņĀüņØś ORR ĒÖ£ņä▒ņØä Ļ░ĆņĪīņ£╝ļ®░ ļ╣äĻĄÉņĀü ļåÆņØĆ ņøÉņ×Éļ╣ä(1:4)ļź╝ Ļ░¢ļŖö ĒĢ®ĻĖł ņ┤ēļ¦żņŚÉņä£ ļé«ņĢśļŗż [33].
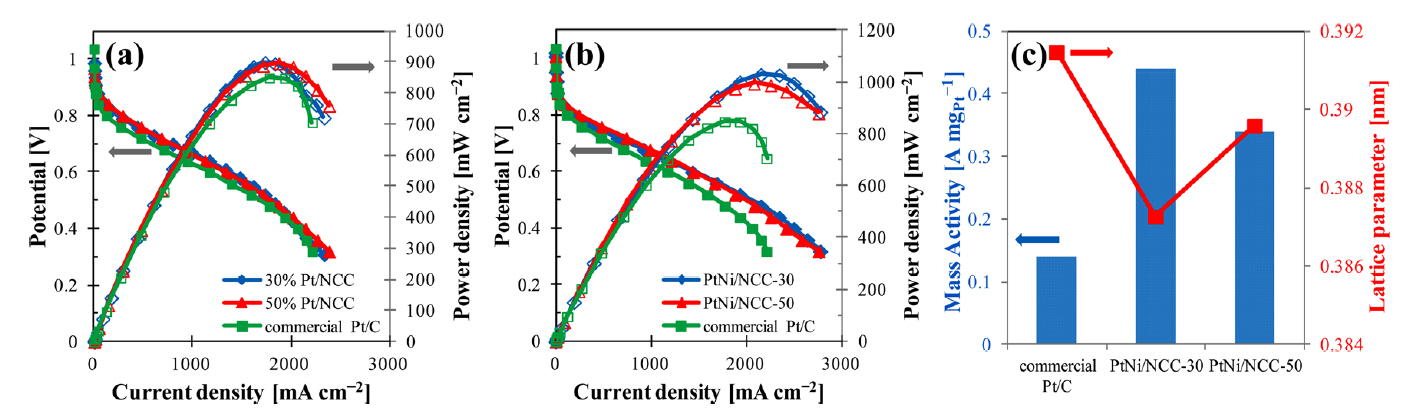
ņĀäĒåĄņĀüņØĖ ĒĢ®ņä▒ļ▓ĢĻ│╝ļŖö ļŗżļźĖ ĒĢ®ņä▒ļ▓ĢņØä ņØ┤ņÜ®ĒĢśņŚ¼ Pt:Ni ņĪ░ņä▒ņØä ļŗżļź┤Ļ▓ī ĒĢ£ ņŚ░ĻĄ¼ļÅä ņ׳ņŚłļŗż [34,35]. Ni-EDA Ēé¼ļĀłņØ┤ĒŖĖ ļ│ĄĒĢ®ņ▓┤ļĪ£ ņĀ£ņ×æĒĢ£ ņ¦Ćņ¦Ćņ▓┤(NCC)ņŚÉ Ptļź╝ ĒĢ©ņ╣©ĒĢśņŚ¼ Ļ░üĻ░üņØś ņĪ░ņä▒ņØ┤ Pt87Ni13, Pt93Ni7ņØĖ ņĮöņ¢┤-ņēś ĻĄ¼ņĪ░ņØś ņ┤ēļ¦ż PtNi/NCC-30, PtNi/NCC-50ņØä ĒĢ®ņä▒ĒĢśņśĆļŗż [34]. ņĀ£ņĪ░ļÉ£ ņ¦Ćņ¦Ćņ▓┤ņØś graphite planeņŚÉ ĒśĢņä▒ļÉ£ pyridinic-NĻ│╝ quaternary-NņØ┤ ORRņŚÉ ņ£Āļ”¼ĒĢ£ ņ×æņÜ®ņØä ĒĢśņśĆņ£╝ļ®░, ĒŖ╣Ē׳ pyridinic-NņØś ĒśĢņä▒ņ£╝ļĪ£ ņ╣┤ļ│ĖļĖöļ×ÖņŚÉ ļ╣äĒĢ┤ NCCļź╝ ņ¦Ćņ¦Ćņ▓┤ļĪ£ ņØ┤ņÜ®ĒĢ£ ņ┤ēļ¦żņØś ĒÖ£ņä▒ņØ┤ ļåÆņĢśļŗż. ĻĘĖļ”╝ 1ņŚÉ ļ│┤ņŚ¼ņ¦ä Ļ▓āĻ│╝ Ļ░ÖņØ┤, PtNi/NCC-30ņØś mass activityļŖö 0.44 A mgPtŌłÆ1ļĪ£, ņāüņÜ® Pt/C ļ│┤ļŗż ņĢĮ 3ļ░░ ļåÆņØĆ ĒÖ£ņä▒ņØä ļ│┤ņśĆļŗż. Magnetron co-sputteringļź╝ ņØ┤ņÜ®ĒĢśņŚ¼ ņØ╝ļ░śņĀüņØĖ ĒÖöĒĢÖņĀü ĒÖśņøÉļ▓Ģ ļ│┤ļŗż Ļ░äļŗ©ĒĢśĻ│Ā ļ╣Āļź┤Ļ▓ī ĒĢ®ĻĖł ņ┤ēļ¦żļź╝ ĒĢ®ņä▒ĒĢśļŖö ņŗ£ļÅäĻ░Ć ņ׳ņŚłļŗż [35]. ņØ┤ ņŚ░ĻĄ¼ņŚÉņä£ magnetronņØś ņĀäļĀźņØä ņĪ░ņĀłĒĢśņŚ¼ ņŚ¼ļ¤¼ ņĪ░ņä▒ņØś PtxNi100ŌłÆx (0 Ōēż x Ōēż 100)ņØä ņĀĢĒÖĢĒĢśĻ▓ī ņĀ£ņ¢┤ĒĢśņśĆĻ│Ā ļæÉĻ╗śņÖĆ Ļ▒░ņ╣ĀĻĖ░ļź╝ ļ╣äņŖĘĒĢ£ ņĀĢļÅäļĪ£ ņĪ░ņĀłĒĢśņśĆļŗż. ļČäņäØ Ļ▓░Ļ│╝ layerņŚÉņä£ņØś Ni ļåŹļÅäĻ░Ć 0-50%ņØ╝ ļĢī ECSAĻ░Ć ņÖäļ¦īĒĢśĻ▓ī ņ”ØĻ░ĆĒĢśļŗżĻ░Ć 50% ņØ┤ņāüņŚÉņä£ ĻĖēĻ▓®ĒĢ£ ņ”ØĻ░Ćļź╝ ļ│┤ņśĆļŗż. ņØ┤Ļ▓āņØĆ NiņØ┤ 50% ņØ┤ņāüņØ╝ ļĢī ņ┤ēļ¦ż Ēæ£ļ®┤ņØś NiĻ░Ć ņÜ®ĒĢ┤ļÉśņ¢┤ Pt-skeleton ĻĄ¼ņĪ░Ļ░Ć ĒśĢņä▒ļÉśĻĖ░ ļĢīļ¼ĖņØ┤ļŗż. Pt-skeleton ĻĄ¼ņĪ░ļŖö Pt-MņŚÉņä£ ņĀäņØ┤ĻĖłņåŹņØ┤ ņÜ®ĒĢ┤ļÉśņ¢┤ ĒśĢņä▒ļÉ£ Pt Ēæ£ļ®┤ņ£╝ļĪ£, Pt-skin ĻĄ¼ņĪ░ņŚÉ ļ╣äĒĢ┤ ORR ĒÖ£ņä▒ņØ┤ ļé«ņØĆ Ļ▓āņ£╝ļĪ£ ņĢīļĀżņĀĖņ׳ļŗż [36]. ĒĢ£ĒÄĖ, ļ¦īļōżņ¢┤ņ¦ä ņāśĒöīļōż ņżæņŚÉņä£ Pt25Ni75ņØś ņä▒ļŖźņØ┤ Ļ░Ćņן ņÜ░ņłśĒĢśņśĆļŗż.
Pt-Ni ņ┤ēļ¦żņØś ļ¬©ņ¢æņØä ņĪ░ņĀłĒĢśņŚ¼ ņ┤ēļ¦ż ĒÖ£ņä▒ņØä Ļ░£ņäĀĒĢśĻ│Āņ×É ĒĢśļŖö ņŚ░ĻĄ¼ļōżņØ┤ ņ׳ņŚłļŗż. Gong ĻĘĖļŻ╣ņØĆ CTAC (cetyl trimethyl ammonium chloride)ņØś ņ¦łļ¤ēĻ│╝ ļ░śņØæņŗ£Ļ░äņØä ņĪ░ņĀłĒĢśņŚ¼ ĻĄÉņ░©ļÉ£ ņØ┤ņżæ ļŹżļ▓© ĒśĢĒā£ņØś ņ┤ēļ¦żļź╝ ĒĢ®ņä▒ĒĢśņśĆļŗż [1]. CTACĻ░Ć ņäĀĒāØņĀüņ£╝ļĪ£ facetņØä cappingĒĢśļŖö Ļ▓āņØä ņØ┤ņÜ®ĒĢśņŚ¼ (100) facetņŚÉ PtĻ│╝ Ni ņøÉņ×ÉĻ░Ć ņ”Øņ░®ļÉśņ¢┤ branched structureļĪ£ ņä▒ņןĒĢśļŖö ļŗżļ®┤ņ▓┤ Pt-Ni ļéśļģĖņŗ£ĒŖĖļź╝ ĒśĢņä▒ĒĢśņśĆļŗż. ņØ┤ņÖĆ Ļ░ÖņØ┤ ĒĢ®ņä▒ļÉ£ PtŌĆōNi cross double dumbbell-like nanostructures (CDDNs)ļŖö ļŗ©Ļ▓░ņĀĢ ļŗżļ®┤ņ▓┤ļĪ£ņŹ©, Ēæ£ļ®┤ ņŚÉļäłņ¦Ćļź╝ ņĄ£ņåīĒÖöĒĢśņŚ¼ ņŚ┤ņŚŁĒĢÖņĀüņ£╝ļĪ£ ļ╣äĻĄÉņĀü ņĢłņĀĢĒĢśņśĆļŗż. ņØ┤ņÖĆ Ļ░ÖņØ┤ ĒĢ®ņä▒ĒĢ£ Pt-Ni ņ┤ēļ¦żņØś mass activityļŖö 3.02 A mgPtŌłÆ1ļĪ£ ņāüņÜ® Pt/Cļ│┤ļŗż 4.2ļ░░, If/Ib ratio (forward / backward peak current density)ļŖö 1.21ļĪ£ 3.5ļ░░ ļåÆņĢä ORRĻ│╝ MOR ņä▒ļŖźņØ┤ ņÜ░ņłśĒ¢łņ£╝ļ®░, CO Ēö╝ļÅģ ļé┤ņä▒ ļśÉĒĢ£ ņāüņÜ® Pt/CņŚÉ ļ╣äĒĢ┤ ņÜ░ņłśĒĢ£ Ļ▓░Ļ│╝ļź╝ ļ│┤ņśĆļŗż. ņØ┤ļ¤¼ĒĢ£ ņä▒ļŖźņŚÉļŖö PtŌĆōNi CDDNsņØś ļŗżņżæ 2ņ░©ņøÉ ļéśļģĖņŗ£ĒŖĖ ĻĄ¼ņĪ░ļĪ£ ņØĖĒĢ£ ļäōņØĆ Ēæ£ļ®┤ņĀüĻ│╝ 3d ņĀäņØ┤ĻĖłņåŹĻ│╝ PtņØś ĒĢ®ĻĖłņØś ņāüņŖ╣ĒÜ©Ļ│╝Ļ░Ć ņśüĒ¢źņØä ņŻ╝ņŚłļŗż. Sievers ņŚ░ĻĄ¼ĒīĆņØĆ ļ▓żņĪ░ņé░ņØä ņØ┤ņÜ®ĒĢśņŚ¼ Ēīöļ®┤ņ▓┤ ĻĄ¼ņĪ░ņØś PtNi/Cļź╝ ĒĢ®ņä▒ĒĢśņśĆļŗż [37]. ņŗżĒŚśņŚÉņä£ ļ▓żņĪ░ņé░ņØś ļåŹļÅä, ļ░śņØæņś©ļÅä, ļ░śņØæņŗ£Ļ░äņØ┤ ņ┤ēļ¦ż ĻĄ¼ņĪ░ņŚÉ ņśüĒ¢źņØä ņŻ╝ņŚłļŗż. ņ┤ēļ¦żĻ░Ć Ēīöļ®┤ņ▓┤ ĻĄ¼ņĪ░ļź╝ ĒśĢņä▒ĒĢśļ®┤ ĒÖ£ņä▒ PtNi(111) ļ®┤ņØä Ēæ£ļ®┤ņŚÉ Ļ░Ćņ¦ĆĻ▓ī ļÉśņ¢┤ ORR ĒÖ£ņä▒ņØ┤ Ē¢źņāüļÉ£ļŗż. ņ┤ēļ¦żĻ░Ć Ēīöļ®┤ņ▓┤ ĻĄ¼ņĪ░ļź╝ ĒśĢņä▒ĒĢśļ®░ ļø░ņ¢┤ļé£ ņ┤ēļ¦ż ĒÖ£ņä▒ņØä Ļ░Ćņ¦ĆĻĖ░ ņ£äĒĢ┤ņä£ļŖö ņĀüņĀłĒĢ£ ļ░śņØæņś©ļÅä(160 ┬░C), ļ░śņØæņŗ£Ļ░ä(12 h), ļ▓żņĪ░ņé░ ļåŹļÅä(10 mM)Ļ░Ć ņżæņÜöĒĢśņśĆņ£╝ļ®░, ĒĢ┤ļŗ╣ņĪ░Ļ▒┤ņŚÉņä£ ĒĢ®ņä▒ļÉ£ ņ┤ēļ¦żņØś Ļ▓ĮņÜ░ mass, specific activityĻ░Ć Pt/Cļ│┤ļŗż Ļ░üĻ░ü 4.7ļ░░, 6.6ļ░░ ļåÆņĢśļŗż.
ņØ┤ņÖĖņŚÉļÅä ĒŖ╣ņĀĢĒĢ£ ņ▓śļ”¼ļź╝ ĒĢśĻ▒░ļéś ņ¦Ćņ¦Ćņ▓┤ļź╝ ļ│ĆĻ▓ĮĒĢśņŚ¼ ņ┤ēļ¦ż ņä▒ļŖźņØä Ļ░£ņäĀĒĢśĻĖ░ļÅä ĒĢśņśĆļŗż. PtņÖĆ NiņØś ļ¬░ļ╣äļź╝ 3:1ļĪ£ ņĪ░ņĀłĒĢśĻ│Ā self-etching(air-etching)Ļ│╝ acid-etchingņØä ĒĢśņŚ¼ Pt-Ni(trace)/GNsļź╝ ĒĢ®ņä▒ĒĢ£ ņŚ░ĻĄ¼ņŚÉņä£, Pt-Ni(trace)/GNsņØś ņä▒ļŖźņØĆ ņŚÉņ╣ŁņØä ĒĢśņ¦Ć ņĢŖņØĆ Pt-Ni/GNsņŚÉ ļ╣äĒĢśņŚ¼ Ļ░£ņäĀļÉśņŚłļŗż [15]. Vulcan XC-72, CNTs, grapheneļź╝ ņé¼ņÜ®ĒĢśņŚ¼ ņ¦Ćņ¦Ćņ▓┤ņŚÉ ļö░ļźĖ Pt1Ni1 ņ┤ēļ¦ż ņä▒ļŖźņØä ņŚ░ĻĄ¼ĒĢ£ Ļ▓░Ļ│╝ņŚÉņä£ļŖö ĻĘĖļלĒĢĆņØä ņ¦Ćņ¦Ćņ▓┤ļĪ£ ņé¼ņÜ®ĒĢ£ ņ┤ēļ¦żĻ░Ć Ļ░Ćņן ņÜ░ņłśĒĢ£ ORR, MOR ņä▒ļŖźņØä ļ│┤ņśĆĻ│Ā Ļ│╝ņé░ĒÖöņłśņåīņØś ņłśņ£©ņØ┤ Ļ░Ćņן ļé«ņĢśļŗż [18]. ņØ┤ļ¤¼ĒĢ£ ņ┤ēļ¦żņØś ņä▒ļŖźņØĆ PtņÖĆ NiņØś ĒĢ®ĻĖłņ£╝ļĪ£ ņØĖĒĢ£ PtņÖĆ Ni ņé░ĒÖöļ¼╝ ņ”ØĻ░ĆļĪ£ ņØĖĒĢ£ Ļ▓āņØ┤ļŗż.
2.2 Pt-Co ĒĢ®ĻĖłņ┤ēļ¦ż
Pt-M ņżæņŚÉņä£ Pt-CoļŖö Pt-FeņÖĆ ļŹöļČłņ¢┤ ļČĆņŗØņä▒Ļ│╝ ņ┤ēļ¦ż ĒÖ£ņä▒ņØ┤ ļåÆņØĆ ĻĘĖļŻ╣ņŚÉ ņåŹĒĢ£ļŗż [27]. Pt-CoļŖö ņé░ņä▒ ļ¦żņ¦łņŚÉņä£ ļåÆņØĆ ORR ĒÖ£ņä▒Ļ│╝ ņĢłņĀĢņä▒ņØä ļ│┤ņØ┤ļ®░, acid-leaching, annealing ļō▒ņØś ņ▓śļ”¼ Ēøä ļ¦īļōżņ¢┤ņ¦ä Pt-Co ņ┤ēļ¦żļōżņØĆ ņ┤ēļ¦ż ĒŖ╣ņä▒Ļ│╝ ņĢłņĀĢņä▒ņØ┤ Ļ░£ņäĀļÉśņŚłļŗż [41,42]. Pt-Co ņĀ£ņĪ░ ņŗ£ CoņØś ņĀäĻĄ¼ņ▓┤ļĪ£ ņé¼ņÜ®ļÉśļŖö ļ¼╝ņ¦łņØĆ ņŻ╝ļĪ£ ņ¦łņé░ ņĮöļ░£ĒŖĖ [2]ņØ┤ļ®░, ņØ┤ņÖĖņŚÉļÅä ņŚ╝ĒÖö ņĮöļ░£ĒŖĖ [27], ņĮöļ░£ĒŖĖ ņĢäņäĖĒģīņØ┤ĒŖĖ [43] ļō▒ņØ┤ ņé¼ņÜ®ļÉśņŚłļŗż.
ņĮöļ░£ĒŖĖ ņĢäņäĖĒģīņØ┤ĒŖĖ, ņŚ╝ĒÖö ņĮöļ░£ĒŖĖ ļō▒ņØś ļŗżļźĖ Co ņĀäĻĄ¼ņ▓┤ļź╝ ņé¼ņÜ®ĒĢśņŚ¼ ņ┤ēļ¦żļź╝ ĒĢ®ņä▒ĒĢśļŖö ņŚ░ĻĄ¼ Ļ▓░Ļ│╝ņŚÉņä£ ņĮöļ░£ĒŖĖ ņĢäņäĖĒģīņØ┤ĒŖĖļź╝ ņé¼ņÜ®ĒĢ£ ņ┤ēļ¦żņØś ORR ņä▒ļŖźņØ┤ ļŹö ņÜ░ņłśĒĢśņśĆļŗż [43]. PtCo/C (C┬░Cl2)ņØś Ļ▓ĮņÜ░ PtCo/C (C4H6CoO4)ļ│┤ļŗż ņ×ģņ×É Ēü¼ĻĖ░Ļ░Ć ņ×æņĢśņ£╝ļéś ORR ņä▒ļŖźņØĆ ļ¢©ņ¢┤ņĪīļŖöļŹ░, ņØ┤ļŖö ClŌĆōĻ░Ć ORR ņä▒ļŖźņŚÉ ļČĆņĀĢņĀüņØĖ ņśüĒ¢źņØä ļ»Ėņ╣śĻĖ░ ļĢīļ¼ĖņØ┤ļŗż.
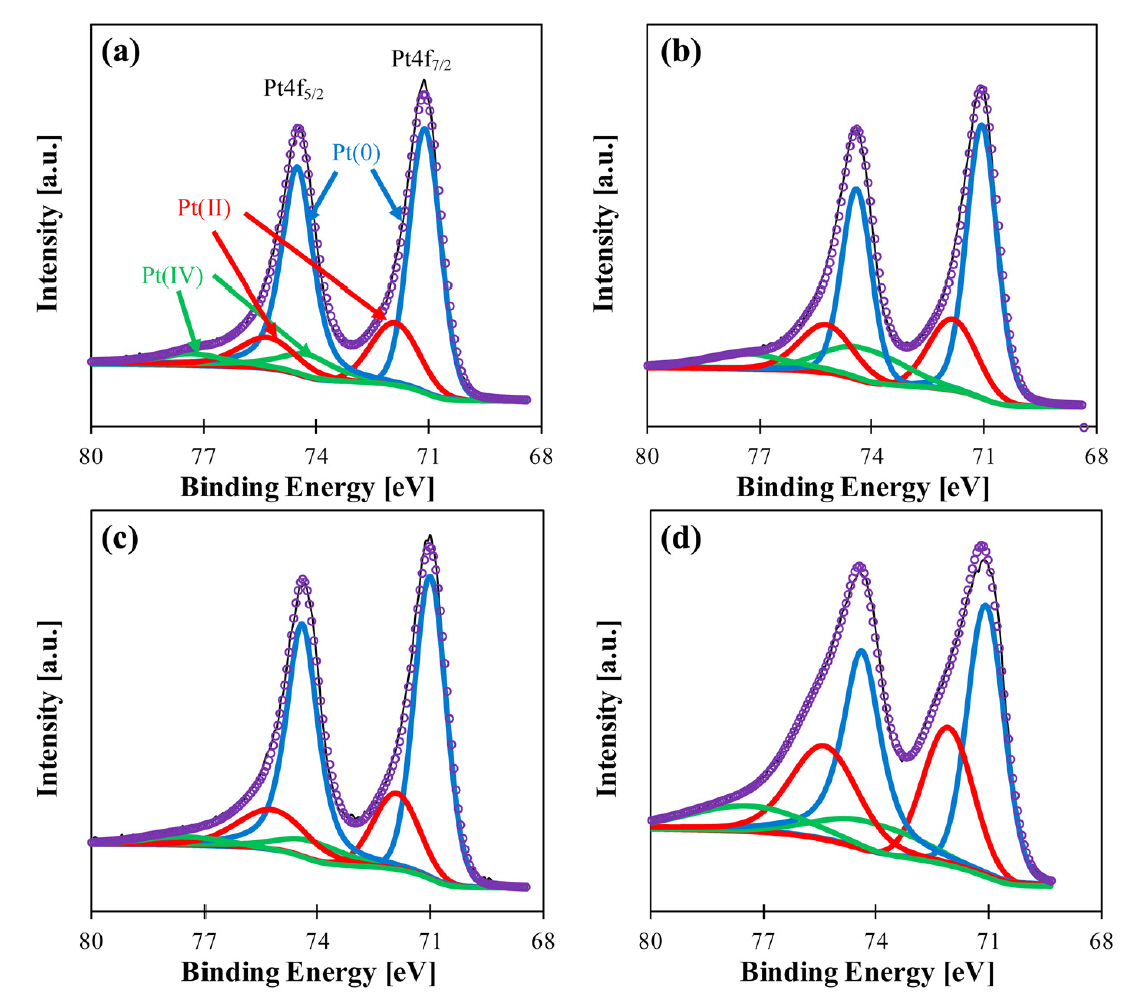
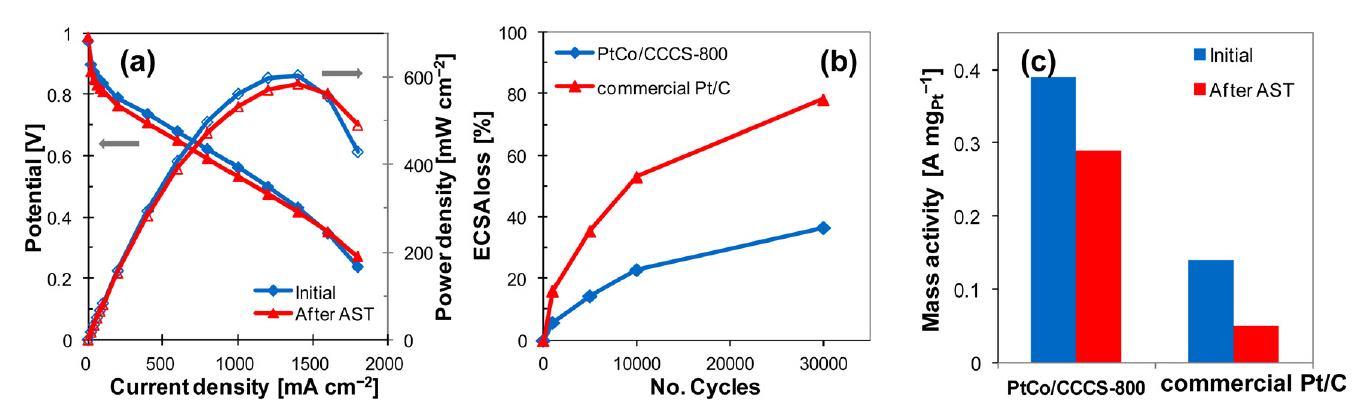
ņé░ ņ▓śļ”¼ ļśÉļŖö ņŚ┤ņ▓śļ”¼Ļ░Ć Pt-Co ņ┤ēļ¦żņŚÉ ļ»Ėņ╣śļŖö ņśüĒ¢źņŚÉ ļīĆĒĢ£ ļ¦ÄņØĆ ņŚ░ĻĄ¼Ļ░Ć ņ׳ņŚłļŗż. ņŚ┤ņ▓śļ”¼ļŖö ņ┤ēļ¦żņØś ĒĢ®ĻĖł ņĀĢļÅä, ņĀäĻĖ░ ņĀäļÅäņä▒, ņĢłņĀĢņä▒ņØä ņ”ØĻ░Ćņŗ£ņ╝░ņ£╝ļéś, Pt ņ×ģņ×ÉĻ░Ć ņØæņ¦æļÉśņ¢┤ ECSAĻ░Ć Ļ░ÉņåīĒĢśņśĆļŗż [27]. 600-900 ┬░CņØś ļŗżļźĖ ņś©ļÅäņŚÉņä£ ņŚ┤ņ▓śļ”¼ļź╝ ĒĢ£ ņŚ░ĻĄ¼ņŚÉņä£ 800 ┬░CņŚÉņä£ ņŚ┤ņ▓śļ”¼ļź╝ ĒĢ£ PtCo/CCCS-800 ĒÖ£ņä▒ņØ┤ Ļ░Ćņן ņÜ░ņłśĒĢśņśĆļŖöļŹ░, ņØ┤ļŖö 800 ┬░CņŚÉņä£ņØś ņŚ┤ņ▓śļ”¼ļĪ£ ņØĖĒĢ┤ ļ®┤ņŗ¼ņĀĢļ░®ņĀĢ ĻĄ¼ņĪ░Ļ░Ć ĒśĢņä▒ļÉśņŚłĻĖ░ ļĢīļ¼ĖņØ┤ļŗż [17]. ļśÉ, ņŚ┤ņ▓śļ”¼ļź╝ 800 ┬░CņŚÉņä£ ņŚ┤ņ▓śļ”¼ ņ¦ĆņåŹ ņŗ£Ļ░äņØ┤ 2 ņŗ£Ļ░äĻ╣īņ¦Ć ņ”ØĻ░ĆĒĢ©ņŚÉ ļö░ļØ╝ ņ┤ēļ¦ż ņä▒ļŖźņŚÉ ĻĖŹņĀĢņĀüņØĖ ņśüĒ¢źņØä ņŻ╝ņŚłļŗż [44]. PtCo/NGCņØś mass activityļŖö ņŚ┤ņ▓śļ”¼ Ēøä 0.18 A mgPtŌłÆ1ņŚÉņä£ 0.45 A mgPtŌłÆ1ļĪ£ ņ”ØĻ░ĆĒĢśņśĆļŗż. ņé░ ņ▓śļ”¼ Ēøä ņŚ┤ņ▓śļ”¼ĒĢ£ ņ┤ēļ¦ż(AL-PtCo/CN), ņé░ ņ▓śļ”¼ĒĢ£ ņ┤ēļ¦ż(L-PtCo/CN), PtCo/CN, ņāüņÜ® Pt/Cļź╝ ļ╣äĻĄÉĒĢ£ Ļ▓░Ļ│╝, AL-PtCo/CNņØ┤ Ļ░ĆņåŹņŖżĒŖĖļĀłņŖżņŗ£ĒŚś ņØ┤Ēøä Ļ░Ćņן ņÜ░ņłśĒĢ£ ņĢłņĀĢņä▒ņØä ļ│┤ņśĆļŗż [45]. ņØ┤ļŖö ORRņŚÉ ļåÆņØĆ ļ░śņØæņä▒Ļ│╝ ņĀäĻĖ░ĒÖöĒĢÖņĀü ņÜ®ņČ£ņŚÉ ļåÆņØĆ ņĀĆĒĢŁņä▒ņØä ļ│┤ņØ┤ļŖö Pt(0)ņØś ĒĢ©ļ¤ēņŚÉ ļö░ļźĖ ļ│ĆĒÖöļĪ£ ļ│┤ņØĖļŗż. ĻĘĖļ”╝ 2ņŚÉ ļéśĒāĆļéĖ Ļ▓āĻ│╝ Ļ░ÖņØ┤, ņ┤łĻĖ░ PtCo ĒĢ®ĻĖłņØś Pt(0)ļŖö 68%ņŚÉ ņØ┤ļź┤ļĀĆņ£╝ļéś ņé░ ņ▓śļ”¼ Ļ│╝ņĀĢņØä Ļ▒░ņ╣£ Ēøä ņĢĮ 59%ņØś ļé«ņØĆ Pt(0) ĒĢ©ļ¤ēņØä ļ│┤ņśĆļŗż. ņØ┤Ēøä ņ×¼ ņŚ┤ņ▓śļ”¼ļź╝ ĒåĄĒĢ┤ 69%ļĪ£ ĒÜīļ│ĄĒĢ£ļŗż. ļ¬©ļōĀ Pt-Co ņ┤ēļ¦żļōżņØĆ Ļ░ĆņåŹņŖżĒŖĖļĀłņŖżņŗ£ĒŚś ņØ┤Ēøä ņāüņÜ® Pt/Cļ│┤ļŗż ņĢłņĀĢņĀüņØ┤ņŚłņ£╝ļéś L-PtCo/CNļŖö Pt(0) ļ╣äņ£© Ļ░Éņåī, Ēāäņåī ņ¦Ćņ¦Ćņ▓┤ ļČĆņŗØ, ĒÖ£ņä▒ļ¼╝ņ¦łĻ│╝ ņ¦Ćņ¦Ćņ▓┤ Ļ▓░ĒĢ®ļĀźņØś Ļ░ÉņåīļĪ£ ņØ┤ļ¤¼ĒĢ£ Ļ▓░Ļ│╝ļź╝ ļ│┤ņśĆļŗż. ņé░ ņ▓śļ”¼ņÖĆ ņŚ┤ņ▓śļ”¼ĒĢ£ ņ┤ēļ¦ż, ņŚ┤ņ▓śļ”¼ĒĢ£ ņ┤ēļ¦ż, ņé░ņ▓śļ”¼ĒĢ£ ņ┤ēļ¦żļź╝ ļ╣äĻĄÉĒĢ£ ņŚ░ĻĄ¼ņŚÉņä£ļŖö ņé░ ņ▓śļ”¼ļź╝ ĒĢśņ¦Ć ņĢŖĻ▒░ļéś ņŚ┤ņ▓śļ”¼ļź╝ Ļ▒░ņ╣£ ņāśĒöīņØĆ ņ┤ēļ¦ż ņ×ģņ×É Ēü¼ĻĖ░Ļ░Ć ņ╗żņ¦ä Ļ▓░Ļ│╝ļĪ£ ņØĖĒĢ┤ ECSAĻ░Ć Ļ░ÉņåīĒĢśņśĆļŗż [43]. ļśÉĒĢ£, ņé░ ņ▓śļ”¼ļź╝ ĒĢ£ ņ┤ēļ¦żļŖö ņ┤ēļ¦ż Ēæ£ļ®┤ņŚÉņä£ CoĻ░Ć ņÖäņĀäĒ׳ ņĀ£Ļ▒░ļÉśņŚłņ£╝ļéś Co ņøÉņ×ÉĻ░Ć ņ┤ēļ¦żņØś Ļ▓░ņĀĢ ĻĄ¼ņĪ░ņŚÉ ņśüĒ¢źņØä ņŻ╝ņ¢┤ ņóŗņØĆ ņä▒ļŖźņØä ļ│┤ņśĆļŗż. ņé░ ņ▓śļ”¼ņÖĆ ņŚ┤ņ▓śļ”¼ļź╝ ļ¬©ļæÉ Ļ▒░ņ╣Ā Ļ▓ĮņÜ░ ņØ┤ņÖĆ Ļ░ÖņØĆ ņ▓śļ”¼ļź╝ ĒĢśņ¦Ć ņĢŖņØĆ ņ┤ēļ¦żļ│┤ļŗż ļåÆņØĆ ĒÖ£ņä▒Ļ│╝ ņĢłņĀĢņä▒ņØä ļ│┤ņØĖļŗż [41].
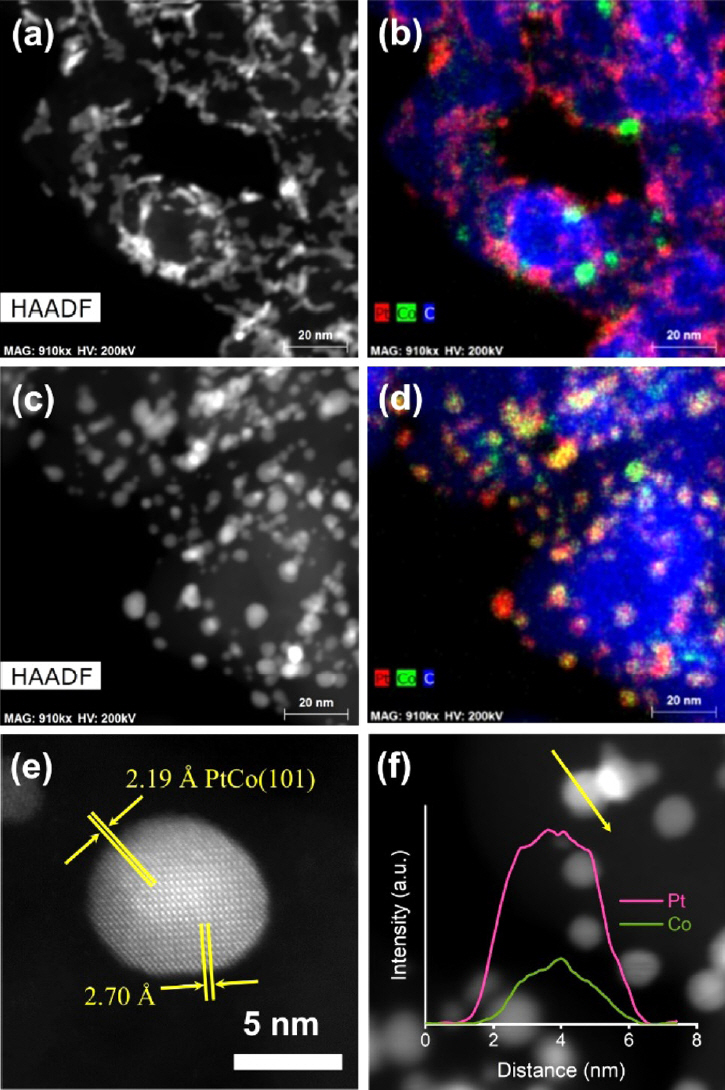
ĻĖ░ņĪ┤ ļ░®ņŗØĻ│╝ļŖö ļŗżļźĖ ĻĖ░ņłĀņØä ļÅäņ×ģĒĢśņŚ¼ Pt-Co ņ┤ēļ¦ż ņä▒ļŖźņØä Ļ░£ņäĀņŗ£ĒéżĻ│Āņ×É ĒĢśļŖö ņŚ░ĻĄ¼ļōżņØ┤ ņ׳ņŚłļŗż. Kumar ĻĘĖļŻ╣ņØĆ ņ┤łņØīĒīī ļ│┤ņĪ░ ĻĖ░ņłĀņØĖ sonochemical ļ░®ļ▓ĢņØä ĒåĄĒĢ┤ ĻĘĀņØ╝ĒĢ£ ņ¦Ćļ”äņØä Ļ░Ćņ¦ä ļéśļģĖ ņ┤ēļ¦żļź╝ ĒĢ®ņä▒ĒĢśņśĆļŗż [2]. Impregnation ļ░®ļ▓Ģņ£╝ļĪ£ ĒĢ®ņä▒ĒĢ£ Pt/C, Pt-Co/CņØś ņ×ģņ×É Ēü¼ĻĖ░Ļ░Ć Ļ░üĻ░ü 7.75 nmņÖĆ 7.39 nmņØĖļŹ░ ļ░śĒĢ┤, sonochemical ļ░®ļ▓Ģņ£╝ļĪ£ ĒĢ®ņä▒ĒĢ£ Pt/C, Pt-Co/C ņ┤ēļ¦żņØś ņ×ģņ×É Ēü¼ĻĖ░ļŖö 5 nm ņØ┤ĒĢśļĪ£ Ļ░ÉņåīĒĢśņśĆļŗż. Pt ĒīīņÜ░ļŹöņÖĆ Coļź╝ high energy ball millingņ£╝ļĪ£ ĒĢ®ĻĖłĒĢśņŚ¼ Co0.75Pt0.25ļź╝ ĒĢ®ņä▒ĒĢ£ ņŚ░ĻĄ¼ņŚÉņä£ ļ│╝ ļ░Ćļ¦üņØä 9ņŗ£Ļ░ä ĒĢ£ Co0.75Pt0.25/C-9h ņ┤ēļ¦żĻ░Ć ļåÆņØĆ ņĢłņĀĢņä▒Ļ│╝ ņ┤ēļ¦żĒÖ£ņä▒ņØä ļ│┤ņśĆļŗż [39]. Co0.75Pt0.25/C-9hņŚÉļŖö CoOņÖĆ ZrOĻ░Ć ņĪ┤ņ×¼Ē¢łņ£╝ļ®░, ZrOļŖö ņ┤ēļ¦żņØś ņĢłņĀĢņä▒ņØä Ļ░£ņäĀĒĢśĻ│Ā Co ņé¼ņÜ®ĒĢśņśĆņØä ļĢī ļ░▒ĻĖł ņøÉņ×ÉņØś ņØæņ¦æņØä ņżäņśĆļŗż. Hu ĻĘĖļŻ╣ņØĆ PtCo nanorod assembles (NRAs)ņŚÉ ņłśņŚ┤ ņ▓śļ”¼ļź╝ ĒĢśņŚ¼ ļüØņØ┤ ļæźĻĖĆĻ│Ā ĻĖĖņØ┤Ļ░Ć ņ¦¦ņØĆ ņ┤łĻĘ╣ļ░Ģ ļéśļģĖļĪ£ļō£ļź╝ ĒĢ®ņä▒ĒĢśņśĆļŗż [46]. ĒāĆ ļģ╝ļ¼ĖļōżņŚÉņä£ ĒĢ®ņä▒ļÉ£ ļéśļģĖ ņÖĆņØ┤ņ¢┤Ļ░Ć low-index facetņØä Ļ░Ćņ¦ĆļŖö Ļ▓āņŚÉ ļ╣äĒĢ┤, NRAsļŖö high-index facetņØä Ļ░Ćņ¦äļŗż. ĻĘĖļ¤¼ļéś, high-index facetņØ┤ ņŚ┤ņŚŁĒĢÖņĀüņ£╝ļĪ£ ļČłņĢłņĀĢĒĢśĻĖ░ ļĢīļ¼ĖņŚÉ ļéśļģĖņÖĆņØ┤ņ¢┤ ļ│ĄĒĢ®ņ▓┤(nanowire assembles, NWAs) ņĢłņŚÉ ņ׳ļŖö grain-boundariesļź╝ ļČĆņŗØņŗ£ņ╝£ ļéśļģĖņÖĆņØ┤ņ¢┤ļź╝ ļüŖņØīņ£╝ļĪ£ņä£ NRAsļź╝ ĒĢ®ņä▒ĒĢśņśĆļŗż. Ļ▓░Ļ│╝ņĀüņ£╝ļĪ£ PtCo NRAs/CļŖö PtCo NWAs/C, Pt NWAs/C, Pt/CņŚÉ ļ╣äĒĢ┤ ļø░ņ¢┤ļé£ ORR ĒÖ£ņä▒ņØä ļ│┤ņśĆļŗż. Batch Ļ│ĄņĀĢņØä ņāØļץĒĢśņŚ¼ ļ¦żņÜ░ ļé«ņØĆ Pt ļ¬░ļ╣ä(Pt:Co = 1:5)ļĪ£ ņ┤ēļ¦żļź╝ ĒĢ®ņä▒ĒĢ£ Ļ▓░Ļ│╝ļÅä ļ│┤Ļ│ĀļÉśņŚłļŗż [41]. ņØ┤ ņŚ░ĻĄ¼ņŚÉņä£ļŖö ĒĢäĒä░ļ¦ü, ņøÉņŗ¼ļČäļ”¼, ņøīņŗ▒, Ļ▒┤ņĪ░, ņŚ┤ņ▓śļ”¼ ļō▒Ļ│╝ Ļ░ÖņØĆ batch Ļ│ĄņĀĢņØä ņāØļץĒĢśļŖö ĒÜ©ņ£©ņĀü ĒĢ®ņä▒ļ▓ĢņØä ņĀ£ņŗ£ĒĢśņśĆļŗż. ļ│╝ ļ░Ćļ¦üņØä ņØ┤ņÜ®ĒĢśņŚ¼ ĒĢ®ņä▒ĒĢ£ PtCo/CļŖö batch Ļ│ĄņĀĢņØä ļÅäņ×ģĒĢ£ ņ┤ēļ¦żņŚÉ ļ╣äĒĢ┤ ļé«ņØĆ ĒÖ£ņä▒ņØä ļ│┤ņśĆņ£╝ļéś in-situ ņĪ░Ļ▒┤ņŚÉņä£ ņāüņÜ® Pt/CņÖĆ ļ╣äņŖĘĒĢśĻ▒░ļéś ļéśņØĆ ņä▒ļŖźņØä ļ│┤ņśĆļŗż. ĻĘĖļ”╝ 3Ļ│╝ Ļ░ÖņØ┤, Pt/C ņ┤ēļ¦żņŚÉ Ļ│╝ļ¤ēņØś ņĀäņØ┤ĻĖłņåŹ ņŚ╝ņØä ĒĢ©ņ╣©ĒĢśļŖö ņØ╝ļ░śņĀüņØĖ ņŗżĒŚśļ▓ĢĻ│╝ ļŗ¼ļ”¼ CoņÖĆ N ņĀäĻĄ¼ņ▓┤ Ēś╝ĒĢ®ļ¼╝ņØä Ēāäņåī ņ¦Ćņ¦Ćņ▓┤ņŚÉ ļ©╝ņĀĆ ļÅäĒĢæĒĢ£ ļÆż Ptļź╝ ļŗ┤ņ¦ĆĒĢśļŖö ņŚ░ĻĄ¼ļōżņØ┤ ņ׳ņŚłļŗż [17,44,47]. NņØś ņ▓©Ļ░ĆļĪ£ ņØĖĒĢ┤ ņŚ┤ņ▓śļ”¼ Ēøä graphite planeņŚÉ pyridinic-NĻ│╝ quaternary-NņØ┤ ĒśĢņä▒ļÉśņŚłĻ│Ā, Pt ņ×ģņ×Éļź╝ ĻĘĀņØ╝ĒĢśĻ▓ī ĒĢ©ņ╣©ņŗ£ņ╝£ ORR ĒÖ£ņä▒ņŚÉ ņśüĒ¢źņØä ņŻ╝ņŚłļŗż. ĻĘĖļ”╝ 4ņØś Ļ▓░Ļ│╝ņÖĆ Ļ░ÖņØ┤ ņØ┤ļ¤¼ĒĢ£ ļ░®ļ▓Ģņ£╝ļĪ£ ĒĢ®ņä▒ļÉ£ ņ┤ēļ¦żļōżņØĆ ĻĘ£ņ╣ÖņĀü ĻĄ¼ņĪ░ļź╝ Ļ░¢ļŖö ļ®┤ņŗ¼ņĀĢļ░®ņĀĢ PtCo ĻĄ¼ņĪ░ļĪ£ ņØĖĒĢ┤ ĻĖłņåŹņØś ņÜ®ĒĢ┤ļź╝ ļ░®ĒĢ┤ĒĢśņŚ¼ ĻĘĖļ”╝ 5ņØś Ļ▓░Ļ│╝ņÖĆ Ļ░ÖņØ┤ Ļ░ĆņåŹņŖżĒŖĖļĀłņŖżņŗ£ĒŚś ņóģļŻī Ēøä ļåÆņØĆ ņĢłņĀĢņä▒ņØä ļ│┤ņśĆļŗż.
2.3 Pt-Fe ĒĢ®ĻĖłņ┤ēļ¦ż
Pt-FeņØĆ ļåÆņØĆ ņ┤ēļ¦ż ĒÖ£ņä▒ņ£╝ļĪ£ ņØĖĒĢ┤ Pt/Cļź╝ ļīĆņ▓┤ĒĢ£ ņ┤ēļ¦żļĪ£ņä£ ņŻ╝ļ¬®ļ░øņĢäņÖöļŗż. ĻĘĖļÅÖņĢł Pt-FeņØś Fe ņĀäĻĄ¼ņ▓┤ļĪ£ņä£ ņ¦łņé░ņ▓ĀņØ┤ Ļ░Ćņן ļäÉļ”¼ ņé¼ņÜ®ļÉśņ¢┤ņÖöņ£╝ļ®░ [48] ņ¦łņé░ņ▓Ā ņÖĖņŚÉļÅä ņŚ╝ĒÖöņ▓Ā [49], ĒÖ®ņé░ņ▓Ā [50], ĒÄśļĪ£ņä╝ [51] ļō▒ņØś ņĀäĻĄ¼ņ▓┤ļź╝ ņé¼ņÜ®ĒĢ£ ņŚ░ĻĄ¼ļōżņØ┤ ņ׳ļŗż.
ņ▓Ā ņĀäĻĄ¼ņ▓┤, ņŚ┤ņ▓śļ”¼ ņĪ░Ļ▒┤, PtņÖĆ FeņØś ļ╣äņ£©ņØ┤ ņ┤ēļ¦żņŚÉ ļ»Ėņ╣śļŖö ņśüĒ¢źņØä ņĪ░ņé¼ĒĢ£ ņŚ░ĻĄ¼ļōżņØ┤ ņ׳ņŚłļŗż. ņ¦łņé░ņ▓Ā ļśÉļŖö ņŚ┤ ņĢłņĀĢņä▒ņØ┤ ļåÆņØĆ ĻĄ¼ņŚ░ņé░ ņ▓Ā ņĢöļ¬©ļŖäņØä Fe ņĀäĻĄ¼ņ▓┤ļĪ£ ņé¼ņÜ®ĒĢśņŚ¼ Ļ░üĻ░ü PtFe/C-N, PtFe/C-Aļź╝ ĒĢ®ņä▒ĒĢ£ Ļ▓░Ļ│╝, PtFe/C-AļŖö PtFe/C-Nļ│┤ļŗż ņ┤ēļ¦ż ĒÖ£ņä▒Ļ│╝ ņĢłņĀĢņä▒ņØ┤ Ēü¼Ļ▓ī Ļ░£ņäĀļÉśņŚłļŗż [48]. Liu ļō▒ņØĆ ņ╣┤ļ│Ė ļéśļģĖ ņä¼ņ£Ā ņ¦Ćņ¦Ćņ▓┤ņŚÉ Pt-Feļź╝ ļŗ┤ņ¦ĆĒĢ£ FeŌĆōPt/CNFs ņ┤ēļ¦żļŖö ĒĢ®ņä▒ ņŗ£ ņŚ┤ņ▓śļ”¼ ņś©ļÅäļź╝ 500-1000 ┬░CļĪ£ ņĪ░ņĀĢĒĢśņśĆļŗż [51]. ļ¦īļōżņ¢┤ņ¦ä ņāśĒöī ņżæņŚÉņä£ 900 ┬░CņŚÉņä£ ņŚ┤ņ▓śļ”¼ĒĢ£ ņāśĒöīņØĖ FeŌĆōPt/CNFs-900ņØś ņ┤ēļ¦ż ņä▒ļŖźņØ┤ Ļ░Ćņן ņÜ░ņłśĒĢśņśĆņ£╝ļ®░ Ļ│╝ņé░ĒÖöņłśņåī ņłśņ£© ļśÉĒĢ£ ļé«ņĢśļŗż. ļśÉ ļŗżļźĖ ņŚ░ĻĄ¼ĻĘĖļŻ╣ņŚÉņä£ļŖö Pt1Fex(x = 1, 2, 3) ņ┤ēļ¦ż ĒĢ®ņä▒ĒĢĀ ļĢī, ņ┤ēļ¦ż Ēæ£ļ®┤ ņĪ░ņä▒ņŚÉ ļö░ļźĖ ņŚ┤ņ▓śļ”¼Ļ░Ć ORRņŚÉ ļ»Ėņ╣śļŖö ņśüĒ¢źņØä ņŚ░ĻĄ¼ĒĢśņśĆļŗż [49]. ņŚ┤ņ▓śļ”¼ļź╝ ĒĢśņ¦Ć ņĢŖņØĆ ņāśĒöīļōżņØś Ļ▓ĮņÜ░ Hupd ņśüņŚŁņØś ņĀäļźśļ░ĆļÅäĻ░Ć Pt1Fe3> Pt1Fe2 > Pt1Fe1 ņł£ņä£ļīĆļĪ£ ņ”ØĻ░ĆĒĢ£ ļ░śļ®┤, ņŚ┤ņ▓śļ”¼ Ēøä ORR ĒÖ£ņä▒ņØĆ Pt1Fe2 > Pt1Fe3 > Pt1Fe1 ņł£ņ£╝ļĪ£ Pt1Fe2ņØś ĒÖ£ņä▒ņØ┤ Ļ░Ćņן ļåÆņĢśļŗż. ņŚÉĒŗĖļĀī ĻĖĆļØ╝ņØ┤ņĮ£ ĒÖśņøÉļ▓ĢņØä ĒåĄĒĢ┤ PtņÖĆ FeņØä ļŗżĻ│Ąņä▒ Ēāäņåī ļéśļģĖņä¼ņ£Ā(porous carbon nanofiber, PCNF)ņŚÉ ļŗ┤ņ¦ĆĒĢ£ Pt4.8Fe/PCNF, Pt9.4Fe/PCNF, Pt6.1Fe/PCNFļź╝ pH7, pH9, pH11ņŚÉņä£ Ļ░üĻ░ü ĒĢ®ņä▒ĒĢ£ ņŚ░ĻĄ¼ļÅä ņ׳ņŚłļŗż [22]. PCNFļŖö ņĀäĻĖ░ļ░®ņé¼ļ▓ĢņØä ĒåĄĒĢ┤ ļŗżņ¢æĒĢ£ Ēü¼ĻĖ░ņØś ĻĖ░Ļ│Ą, Ļ▒░ņ╣£ Ēæ£ļ®┤, ļ¦ÄņØĆ Ļ▓░ĒĢ©ņØä Ļ░ĆņĪīļŗż. ņ┤ēļ¦żļōż ņżæ Pt4.8Fe/PCNFņØś Ļ░£ņŗ£ņĀäņ£äņÖĆ ļ░śĒīīņĀäņ£äļŖö Ļ░üĻ░ü 0.978, 0.824 VļĪ£, Pt/CņØś Ļ░Æ(0.96, 0.822 V)ļ│┤ļŗż ļåÆņØĆ ņä▒ļŖźņØä ļ│┤ņśĆļŗż. Wu ĻĘĖļŻ╣ņØĆ PtĻ│╝ Pt3Fe, PtFe3ņØä ļ╣äĻĄÉĒĢśņŚ¼ ļČäņäØĒĢśņŚ¼, PtņŚÉ ļ╣äĒĢ┤ Pt-FeņØś ĒÖ£ņä▒ņØ┤ ļåÆņØĆ Ļ▓āņØä ĒÖĢņØĖĒĢśņśĆļŗż [52]. Pt skin Ēæ£ļ®┤Ļ│╝ ĒØĪņ░®ļ¼╝(adsorbate) Ļ░äņØś ņāüĒśĖņ×æņÜ®ņØĆ Pt(111) > Pt-skinned Pt3Fe (111) > Pt-skinned PtFe3(111) ņł£ņ£╝ļĪ£ Ļ░ĢĒ¢łļŗż. ņé░ņåīĻ░Ć ĒØĪņ░®ĒĢśĻ│Ā ļ¼╝ ļČäņ×ÉĻ░Ć Ēāłņ░®ĒĢśļŖö 4ņĀäņ×É Ļ│╝ņĀĢņŚÉņä£, Pt-skinned PtFe3(111)ļŖö ņé░ņåī ĒØĪņ░®ņŚÉ ļīĆĒĢ┤ Ļ░Ćņן ļé«ņØĆ ĒÖ£ņä▒ĒÖö ņŚÉļäłņ¦Ćļź╝ ĒĢäņÜöļĪ£ Ē¢łĻ│Ā, ļ¼╝ ļČäņ×É Ēāłņ░®ņŚÉ ĒĢäņÜöĒĢ£ ņŚÉļäłņ¦Ć Ļ░ÆņØ┤ Ļ░Ćņן ļé«ņĢśļŗż. ņØ┤ļĪ£ ņØĖĒĢ┤, ļ¼╝ ļČäņ×ÉĻ░Ć Pt-skinned PtFe3(111) Ēæ£ļ®┤ņŚÉņä£ ņēĮĻ▓ī Ēāłņ░®ļÉśņ¢┤ ļŗżņØī ļ░śņØæņØä ļ╣Āļź┤Ļ▓ī ņØ┤ņ¢┤ļéśĻ░ł ņłś ņ׳ļŗż. Ļ▓░ļĪĀņĀüņ£╝ļĪ£ Fe ĒĢ©ļ¤ēņØ┤ ļŹö ļåÆņĢä ĒśĢņä▒ļÉśļŖö Pt-skinned PtFe3(111)ņØ┤ Pt-skinned Pt3Fe(111)ļ│┤ļŗż ļåÆņØĆ ĒÖ£ņä▒ņØä ļ│┤ņśĆļŗż. Ēāäņåīļź╝ PtFe ĒĢ®ĻĖł ņ┤ēļ¦żņŚÉ ņĮöĒīģĒĢśņŚ¼ ORRņŚÉ ļīĆĒĢ£ ļé┤ĻĄ¼ņä▒ņØä Ē¢źņāüņŗ£ĒéżļŖö ļģĖļĀźļÅä ļ│┤Ļ│ĀļÉśņŚłļŗż [53]. ĒāäņåīĻ░Ć ĒÆŹļČĆĒĢ£ ļ”¼Ļ░äļō£ļź╝ ĒĢ©ņ£ĀĒĢ£ ĻĖłņåŹ ņĀäĻĄ¼ņ▓┤ļź╝ ņé¼ņÜ®ĒĢśņŚ¼ ĒĢ®ĻĖł ņ┤ēļ¦żļź╝ ĒĢ®ņä▒ĒĢśļ®░, ĻĘĖļ”╝ 6Ļ│╝ Ļ░ÖņØ┤ ļÅÖņŗ£ņŚÉ ņ¢ćņØĆ ĒāäņåīņĖĄņØ┤ ĒśĢņä▒ļÉśļŖö ĒśäņāüņØä ņ£ĀļÅäĒĢśņŚ¼ ņ┤ēļ¦żļź╝ ņĀ£ņĪ░ĒĢśņśĆļŗż. ļé┤ĻĄ¼ņä▒ ņŗ£ĒŚś ĒøäņŚÉ ņāüņÜ® Pt/CņØś ļ░śĒīī ņĀäņ£äĻ░ÆņØ┤ ņĢĮ 70 mV Ļ░ÉņåīĒĢ£ļŹ░ ļ░śĒĢ┤, ĒāäņåīņĖĄņØä ņĀüņÜ®ĒĢ£ ĒĢ®ĻĖł ņ┤ēļ¦żļŖö ņĢĮ 20-30 mV Ļ░ÉņåīĒĢ£ Ļ▓āņØä ĒÖĢņØĖĒĢĀ ņłś ņ׳ļŗż. ņāüņÜ® Pt/CņØś ECSAĻ░Ć ņĢĮ 60% Ļ░ÉņåīĒĢ£ Ļ▓āĻ│╝ ļŗ¼ļ”¼, ĒĢ®ĻĖł ņ┤ēļ¦żļŖö ļ¼┤ņŗ£ĒĢĀ ļ¦īĒĢ£ ļ│ĆĒÖöļ¦īņØ┤ Ļ┤ĆņĖĪļÉśņŚłļŗż.
ĻĘĖ ņÖĖņŚÉļÅä ņ£ĪĻ░üĒśĢņØś nanoplate Pt-M (M= Fe, Co, Ni)ļź╝ Ļ░äļŗ©ĒĢ£ ļ░®ļ▓Ģņ£╝ļĪ£ ņĀ£ņĪ░ĒĢśĻĖ░ ņ£äĒĢ£ ņŚ░ĻĄ¼Ļ░Ć ņ׳ņŚłļŗż [40]. ĒĢ®ņä▒ļÉ£ PtFe ņ┤ēļ¦żļŖö ņĢĮ 2.6 nmņØś ņ£ĪĻ░üĒśĢ nanoplate ĻĄ¼ņĪ░ļź╝ Ļ░Ćņ¦ĆĻ│Ā ņ׳ņŚłņ£╝ļ®░ Pt, PtCo, PtNiĻ│╝ ļ╣äĻĄÉĒĢśņŚ¼ ņé░ņä▒, ņŚ╝ĻĖ░ņä▒ ņĀäĒĢ┤ņ¦łņŚÉņä£ ņÜ░ņłśĒĢ£ ņ┤ēļ¦ż ĒÖ£ņä▒ņØä ļ│┤ņśĆļŗż. ņŗżĒŚśņŚÉņä£ PtM ĒĢ®ņä▒ ņŗ£ Ēżļ”äņé░ņŚÉ ĻĖłņåŹ ņĀäĻĄ¼ņ▓┤ņØĖ ĻĖłņåŹņŚ╝ĒÖöļ¼╝ņØä ņÜ®ĒĢ┤ņŗ£ņ╝░ļŖöļŹ░, Ēżļ”äņé░ņØ┤ Ļ│╝ņĀäņ£äļź╝ ļé«Ļ▓ī ņ£Āņ¦ĆĒĢśĻ│Ā ņ┤ēļ¦ż Ēæ£ļ®┤ņŚÉ ņżæĻ░äņ▓┤ ņČĢņĀüņØä Ļ░Éņåīņŗ£ĒéżļŖö ĒÜ©Ļ│╝Ļ░Ć ņ׳ļŗżļŖö Ļ▓āņØ┤ ņØ┤ņĀä ņŚ░ĻĄ¼ [54]ņŚÉņä£ ņ×ģņ”ØļÉ£ ļ░ö ņ׳ļŗż. ĒøäĻĖ░ ņĀäņØ┤ ĻĖłņåŹņØĆ PtņŚÉ ļ╣äĒĢ┤ 5d vacancyņØä ļŹö ļ¦ÄņØ┤ Ļ░Ćņ¦ĆĻ│Ā ņ׳ļŗż. ņØ┤ļ¤¼ĒĢ£ vacancyļŖö O2 ĒØĪņ░®ņØä ļÅĢĻ│Ā O-O Ļ▓░ĒĢ®ņØĆ ņĢĮĒĢśĻ▓ī ĒĢśņŚ¼ O2 ļ░śņØæņåŹļÅäļź╝ ļåÆņØĖļŗż. FeļŖö Co, NiĻ│╝ Ļ░ÖņØĆ ĒøäĻĖ░ ņĀäņØ┤ ĻĖłņåŹņŚÉ ļ╣äĒĢ┤ 5d vacancyĻ░Ć ļŹö ļ¦ÄĻĖ░ ļĢīļ¼ĖņŚÉ ņ┤ēļ¦ż ņä▒ļŖźņØ┤ ļåÆņØĆ Ļ▓āņ£╝ļĪ£ ņČöņĀĢļÉ£ļŗż.
2.4 ĻĘĖņÖĖ Pt-M ĒĢ®ĻĖłņ┤ēļ¦ż
ņĢ×ņäĀ Pt-Ni, Pt-Co, Pt-Fe ņÖĖņŚÉļÅä Pt-Sn [38,55,56], Pt-Cu [57,58], Pt-Pd [59,60], Pt-Ag [61,62], Pt-Cr [63], Pt-Mn [64], Pt-Au [65], Pt-B [66] ļō▒ņØś ļ░öņØ┤ļ®öĒāł ņ┤ēļ¦żĻ░Ć ņŚ░ĻĄ¼ļÉśņŚłļŗż. ļ¦ÄņØĆ ņŚ░ĻĄ¼ņŚÉņä£ PtņÖĆ MņØś ņĀüņĀłĒĢ£ ļ╣äņ£©ņØä ņ░ŠĻ▒░ļéś [56,60,62,63,66] ņ¦Ćņ¦Ćņ▓┤ [55,58,59,65,66] ļśÉļŖö ņŚ┤ņ▓śļ”¼ ņĪ░Ļ▒┤ [63]ņØä ļŗ¼ļ”¼ĒĢśļŖö ļō▒ņØś ļ░®ļ▓Ģņ£╝ļĪ£ ņ┤ēļ¦ż ņä▒ļŖźņØä Ļ░£ņäĀĒĢśņśĆļŗż.
Beyhan ĻĘĖļŻ╣ņØś ņŚ░ĻĄ¼ņŚÉ ļö░ļź┤ļ®┤, Pt-Sn/CņØĆ Pt-Ni/C, Pt-Sn-Ni/CņŚÉ ļ╣äĒĢ┤ ņĀäĻĖ░ĒÖöĒĢÖņĀü ņä▒ļŖźņØ┤ ņÜ░ņłśĒ¢łļŗż [38]. ņØ┤ļŖö Pt-Sn/CņŚÉņä£ ņĀüņĀłĒĢ£ ņ¢æņ£╝ļĪ£ ņĪ┤ņ×¼ĒĢśļŖö SnO2ļĪ£ ņØĖĒĢ┤ ļ¼╝Ļ│╝ ņé░ņåīņØś ĒØĪņ░®ņØ┤ ORRņŚÉ ņ£Āļ”¼ĒĢśĻ▓ī ņ×æņÜ®Ē¢łĻĖ░ ļĢīļ¼ĖņØ┤ļŗż. 1-D Ēāäņåī ļéśļģĖĒŖ£ļĖīņÖĆ 2-D ĻĘĖļלĒĢĆ ĒĢśņØ┤ļĖīļ”¼ļō£ Ēāäņåī heterostructureļź╝ ņØ┤ņÜ®ĒĢśņŚ¼ ļåÆņØĆ Ēæ£ļ®┤ņĀüĻ│╝ ņĀäĻĖ░ņĀäļÅäņä▒ņØ┤ ņÜ░ņłśĒĢ£ ņ¦Ćņ¦Ćņ▓┤ļź╝ ņé¼ņÜ®ĒĢ£ Pt-Sn/PCNTņŚÉ ļīĆĒĢ£ ņŚ░ĻĄ¼ Ļ▓░Ļ│╝Ļ░Ć ņåīĻ░£ļÉśņŚłļŗż [55]. PCNTļŖö ņ╣┤ļ│Ė ļéśļģĖ ĒŖ£ļĖīņŚÉ ļŹö ļ¦ÄņØĆ Ļ▓░ĒĢ©ņØä ļ¦īļōżņŚłĻ│Ā, ņØ┤ļ¤¼ĒĢ£ ņä▒ņ¦łņØĆ ņ╣┤ļ│Ė ļéśļģĖĒŖ£ļĖī Ļ│Āņ£ĀņØś ņןņĀÉĻ│╝ ļŹöļČłņ¢┤ ļ¦ÄņØĆ anchoring sitesļź╝ ņāØņä▒ĒĢśņŚ¼ ņ┤ēļ¦ż ņØ┤ņÜ®ļźĀņØä Ē¢źņāüņŗ£ņ╝░ļŗż. ļśÉĒĢ£ Pt-Sn/PCNTļŖö Pt-Sn/CNTņŚÉ ļ╣äĒĢ┤ ņŚÉĒāäņś¼ ņé░ĒÖö ļ░śņØæņŚÉņä£ņØś ņżæĻ░ä ņé░ļ¼╝ņØś ņ¢æņØ┤ ļŹö ņĀüņŚłļŗż. PtņÖĆ SnņØś ņ¦łļ¤ēļ╣äļź╝ 80/20(PtSn-A), 65/35(PtSn-B), 50/50(PtSn-C)ļĪ£ ļŗżļź┤Ļ▓ī ĒĢśņŚ¼ ļ╣äĻĄÉĒĢśļŖö ņŚ░ĻĄ¼Ļ░Ć ļ│┤Ļ│Ā ļÉśņŚłļŗż [56]. Sn ĒĢ®ĻĖł ņŗ£ Sn ņé░ņåīĒĢ©ņ£ĀņóģņØś ĒśĢņä▒ņ£╝ļĪ£ ņé░ĒÖöļź╝ ņ¢ĄņĀ£ĒĢĀ ņłś ņ׳Ļ│Ā ņØĖņĀæĒĢ£ PtņØś unoccupied d-bandļź╝ ļé«ņČ£ ņłś ņ׳ņŚłļŗż. SnņØś ĒĢ©ļ¤ēņØ┤ Ļ░Ćņן ļåÆņØĆ PtSn-CņØś ņ┤ēļ¦ż ĒÖ£ņä▒ņØ┤ Ļ░Ćņן ņÜ░ņłśĒĢśņśĆņ£╝ļéś, ņØīņĀäĒĢśņØś Sn ņé░ņåīĒĢ©ņ£ĀņóģņØ┤ ņ¢æņĀäĒĢśņØś Ptļź╝ ļüīņ¢┤ļŗ╣Ļ▓© Pt ļéśļģĖ ļĪ£ļō£ ĻĄ¼ņĪ░ļź╝ ĒīīĻ┤┤ĒĢśņŚ¼ ļé┤ĻĄ¼ņä▒ ņŗ£ĒŚśņŚÉņä£ ņĢłņĀĢņä▒ņØ┤ Ļ░Ćņן ļé«ņĢśļŗż. ļ░śļ®┤ņŚÉ, PtSn-AņŚÉņä£ 1-D Pt ļéśļģĖ ņÖĆņØ┤ņ¢┤ņØś ļåŹļÅäĻ░Ć Ļ░Ćņן ļåÆņĢśĻ│Ā ļé┤ĻĄ¼ņä▒ ņŗ£ĒŚśņŚÉņä£ Ļ░Ćņן ņÜ░ņłśĒĢ£ ņĢłņĀĢņä▒ņØä ļ│┤ņśĆļŗż.
ļŗżņ¢æĒĢ£ ņ¦Ćņ¦Ćņ▓┤ļź╝ ņé¼ņÜ®ĒĢśņŚ¼ Pt-Cu ņ┤ēļ¦żļź╝ ĒĢ®ņä▒ĒĢ£ ņŚ░ĻĄ¼Ļ░Ć ļ│┤Ļ│ĀļÉśņŚłļŗż [58]. ņŚ░ĻĄ¼ņŚÉ ņé¼ņÜ®ļÉ£ ņ¦Ćņ¦Ćņ▓┤ļŖö ņ╣┤ļ│Ė ļéśļģĖ ņä¼ņ£Ā(CNFs), ļŗżņżæļ▓Į ĒāäņåīļéśļģĖĒŖ£ļĖī(MWCNTs), Vulcan XC72 ĒāäņåīņØ┤ļŗż. ĒĢ®ņä▒ļÉ£ ņ┤ēļ¦żļōżņØĆ Pt-Cu/CņÖĆ Pt/Cļ│┤ļŗż ņÜ░ņłśĒĢ£ CO ļé┤ņä▒ņØä Ļ░ĆņĪīņ£╝ļ®░, ņØ┤ļōż ņżæ CNFņÖĆ MWCNTņŚÉ ļŗ┤ņ¦ĆĒĢ£ Pt-Cu ĒĢ®ĻĖł ņ┤ēļ¦żĻ░Ć Ļ░üĻ░ü Ļ░Ćņן ļåÆņØĆ mass activityņÖĆ specific activityļź╝ ļ│┤ņśĆļŗż. ĻĘĖļ”¼Ļ│Ā ļæÉ ņ┤ēļ¦żļŖö PtņØś ņ¢æņØä 50% ņżäņŚ¼ ņé¼ņÜ®Ē¢łņØä Ļ▓ĮņÜ░ņŚÉļÅä ļåÆņØĆ ņĢłņĀĢņä▒ņØä ļ│┤ņśĆļŗż. Geboes ĻĘĖļŻ╣ņØĆ Ļ░łļ░öļŗē Ļ│ĄņĀĢņØä ĒżĒĢ©ĒĢ£ Ļ░äļŗ©ĒĢ£ 2 ļŗ©Ļ│ä ņŗżĒŚśļ▓Ģņ£╝ļĪ£ Pt-Cu/Cļź╝ ĒĢ®ņä▒Ē¢łļŗż [57]. Pt-CuņØś ļé«ņØĆ ļČäņé░ļÅäļĪ£ ņØĖĒĢ┤ Pt-Cu/CņØś ECSAĻ░ÆņØĆ ņāüņÜ® Pt/CņØś Ļ░Æļ│┤ļŗż ļé«ņĢśņ£╝ļéś ļ”¼Ļ░äļō£ ĒÜ©Ļ│╝ļĪ£ ņØĖĒĢ┤ specific activityļŖö ņāüņÜ® Pt/C ļ│┤ļŗż ļåÆņĢśļŗż.
CNTņØś ļČäņé░ļÅäĻ░Ć ņ┤ēļ¦żņŚÉ ļ»Ėņ╣śļŖö ņśüĒ¢źņØä ņŚ░ĻĄ¼ĒĢśĻĖ░ ņ£äĒĢ┤ ņØīņØ┤ņś©ņä▒ Ļ│äļ®┤ĒÖ£ņä▒ņĀ£ņØĖ ļÅäļŹ░ņŗż ĒÖ®ņé░ ļéśĒŖĖļź©(SDS)ņÖĆ ņ¢æņØ┤ņś©ņä▒ Ļ│äļ®┤ĒÖ£ņä▒ņĀ£ ņäĖĒŖĖļ”¼ļ¬©ļŖä ļĖīļĪ£ļ¦łņØ┤ļō£(CTAB)ļź╝ ņé¼ņÜ®ĒĢśņŚ¼ Pt-Pd/CNT-SņÖĆ Pt-Pd/CNT-Cļź╝ Ļ░üĻ░ü ĒĢ®ņä▒ļÉśņŚłļŗż [59]. Ļ│äļ®┤ĒÖ£ņä▒ņĀ£ļŖö Pt-PdņØś ņ×ģņ×É Ēü¼ĻĖ░ņŚÉļŖö ņśüĒ¢źņØä ļ»Ėņ╣śņ¦Ć ņĢŖņĢśņ£╝ļéś MWCNTņØś ļČäņé░ņŚÉ ņśüĒ¢źņØä ņŻ╝ņŚłļŗż. ļæÉ Ļ│äļ®┤ĒÖ£ņä▒ņĀ£ ņżæņŚÉņä£ ņØīņØ┤ņś©ņä▒ Ļ│äļ®┤ĒÖ£ņä▒ņĀ£ņØś Ļ▓ĮņÜ░, MWCNT ļŗżļ░£ņØä ļČäņé░ņŗ£ĒéżĻ│Ā Pt-Pd ļéśļģĖņ×ģņ×Éļź╝ ĻĘĀņØ╝ĒĢśĻ▓ī ļČäņé░ņŗ£ĒéżļŖö Ļ▓āņØä ļÅäņÖöļŗż. ņ¢æņØ┤ņś©ņä▒ Ļ│äļ®┤ĒÖ£ņä▒ņĀ£ņØś Ļ▓ĮņÜ░ņŚÉļŖö ļ░śļīĆļĪ£ MWCNT ļŗżļ░£Ļ│╝ ņ×ģņ×É ļČäņé░ņŚÉ ļÅäņøĆņØä ņŻ╝ņ¦Ć ņĢŖņĢśļŗż. ņØ┤ļ¤¼ĒĢ£ ņśüĒ¢źņØä ļ░öĒāĢņ£╝ļĪ£ Pt-Pd/CNT-SņØĆ ļåÆņØĆ ņĢłņĀĢņä▒Ļ│╝ ņ┤ēļ¦żĒÖ£ņä▒ņØä ļ│┤ņśĆļŗż. Pt-PdņØś Pt ņ¦łļ¤ēļ╣ä(1-7%)ņÖĆ PBI wt%(10-40 wt%)ļź╝ ņĪ░ņĀĢĒĢśņŚ¼ Pt-Pd/Cļź╝ ĒĢ®ņä▒ĒĢśņśĆņØä ļĢī, PBI 30 wt%ņØ┤Ļ│Ā Pt:PdņØś ņ¦łļ¤ēļ╣äĻ░Ć 5:95ņØĖ ņ┤ēļ¦żĻ░Ć Ļ░Ćņן ņóŗņØĆ ņ┤ēļ¦żĒÖ£ņä▒ņØä ļ│┤ņśĆļŗż [60]. ņ¦Ćļéśņ╣śĻ▓ī ļåÆņØĆ PBI 40 wt%ļŖö ļ¦ÄņØĆ ņ¢æņØś ņżæĒĢ®ņ▓┤ļĪ£ ņØĖĒĢ┤ ļ░śņØæļ¼╝ ņØ┤ļÅÖņØä ņĀĆĒĢ┤ņŗ£ņ╝£ ņ┤ēļ¦ż ĒÖ£ņä▒ņØ┤ Ļ░Éņåīņŗ£ņ╝░ļŗż.
Ruiz-Camacho ĻĘĖļŻ╣ņØĆ sonochemical Ļ│ĄņĀĢņØä ņØ┤ņÜ®ĒĢśņŚ¼ Pt-Ag/CņØä ĒĢ®ņä▒ĒĢśņśĆļŗż [61]. Pt/CņÖĆ Ag/Cļ│┤ļŗż ļø░ņ¢┤ļé£ ņ┤ēļ¦ż ĒÖ£ņä▒ņØä ļ│┤ņśĆņ£╝ļ®░, ņé░ņä▒ ņĀäĒĢ┤ņ¦łņŚÉņä£ Pt/C ORR ņ┤ēļ¦ż ĒÖ£ņä▒ņØś 1.5ļ░░ ļåÆņØĆ ĒÖ£ņä▒ņØä ļ│┤ņśĆļŗż. AgņÖĆ PtņØś ļ╣äņ£©ņØä ņĪ░ņĀĢĒĢśņŚ¼ ņĮöņ¢┤-ņēś ĻĄ¼ņĪ░ņØś Ag@Pt/Cļź╝ ĒĢ®ņä▒ĒĢśņśĆļŗż [62]. ņŗżĒŚśĻ▓░Ļ│╝ Ag:PtņØś ņ¦łļ¤ēļ╣äĻ░Ć 1:1, 1:3, 1:9ņØĖ ņāśĒöīļōż ņżæ 1:3ņØś ņ¦łļ¤ēļ╣äļź╝ Ļ░Ćņ¦ä ņ┤ēļ¦żņØś ņ┤ēļ¦ż ĒÖ£ņä▒ņØ┤ Ļ░Ćņן ļø░ņ¢┤ļé¼ņ£╝ļ®░, ņØ┤ļŖö ņāüņÜ® Pt/CņØś Ļ░£ņŗ£ņĀäņ£ä(0.629 V)ļ│┤ļŗż ļåÆņØĆ Ļ░Æ(0.712 V)ņØä ļ│┤ņśĆļŗż.
PtņÖĆ CrņØś ņøÉņ×Éļ╣äļź╝ 1:1ņŚÉņä£ 5:1Ļ╣īņ¦Ć ņĪ░ņĀłĒĢśĻ│Ā ņŚ┤ņ▓śļ”¼ ņĪ░Ļ▒┤ņØä ļŗ¼ļ”¼ĒĢśņŚ¼ PtCr/Cļź╝ ĒĢ®ņä▒ĒĢ£ ņŚ░ĻĄ¼ņŚÉņä£, PtņØś ņøÉņ×Éļ╣äĻ░Ć ņ”ØĻ░ĆĒĢ©ņŚÉ ļö░ļØ╝ PtCrņØś ĒĢ®ĻĖłĒÖöļÅäļŖö ļé«ņĢäņĪīņ£╝ļ®░ ņØ┤ ņżæ Pt:CrĻ░Ć 3:1ņØĖ ņāśĒöīņØś ņ┤ēļ¦ż ĒÖ£ņä▒ņØ┤ Ļ░Ćņן ļø░ņ¢┤ļé¼ļŗż [63]. ņŚ┤ņ▓śļ”¼ļŖö ņ┤ēļ¦żņØś ņĢłņĀĢņä▒Ļ│╝ ĒĢ®ĻĖłĒÖö ņĀĢļÅä ļō▒ņŚÉ ņśüĒ¢źņØä ņŻ╝ņŚłļŖöļŹ░, ņ¦łņåī ļČäņ£äĻĖ░ņŚÉņä£ ņŚ┤ņ▓śļ”¼ ĒĢ£ ņ┤ēļ¦żņØś Ļ▓ĮņÜ░ ņ┤ēļ¦ż ņŚ┤ņ▓śļ”¼ ĒĢśņ¦Ć ņĢŖņØĆ ņ┤ēļ¦żņŚÉ ļ╣äĒĢ┤ ņĢłņĀĢņä▒Ļ│╝ ĒĢ®ĻĖłĒÖöļÅäĻ░Ć ņ”ØĻ░ĆĒĢśņśĆļŗż.
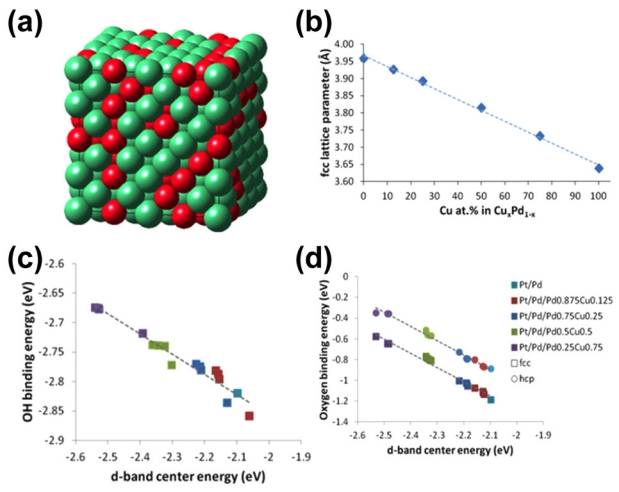
ņēśņØś ņøÉņ×ÉņĖĄ ņłśļź╝ 1, 1.5, 2ļĪ£ ņĪ░ņĀłĒĢśņŚ¼ ņĮöņ¢┤-ņēś ĻĄ¼ņĪ░ņØś Mn@Pt/Cļź╝ ĒĢ®ņä▒ĒĢ£ ņŚ░ĻĄ¼ļÅä ņ׳ņŚłļŗż [64]. CV ļŹ░ņØ┤Ēä░ļź╝ ĒåĄĒĢ┤ Mn ņĮöņ¢┤Ļ░Ć Pt ņēśņØś ņé░ņåī ņ╣£ĒÖöņä▒ņØä ņ”ØĻ░Ćņŗ£ĒéżļŖö Ļ▓āĻ│╝ Pt ĒĢ©ļ¤ēņØ┤ ņ”ØĻ░ĆĒĢĀ ļĢī ņ┤ēļ¦ż Ēæ£ļ®┤ņŚÉ ĒśĢņä▒ļÉ£ ņé░ĒÖöļ¼╝ņØś ņĢłņĀĢņä▒ņØ┤ ņ”ØĻ░ĆĒĢśļŖö Ļ▓āņØä ĒÖĢņØĖĒĢśņśĆļŗż. ļśÉĒĢ£ ļ¦īļōżņ¢┤ņ¦ä ņ┤ēļ¦ż ņżæņŚÉņä£ ņēśņØś ņøÉņ×ÉņĖĄ ņłśĻ░Ć 1.5ņØĖ ņāśĒöīņØś specific activityņÖĆ mass activityĻ░Ć Ļ░Ćņן ļåÆņĢśņ£╝ļ®░, ņēśņØś ņøÉņ×ÉņĖĄ ņłśĻ░Ć 2ņØĖ ņāśĒöīņØś ņĢłņĀĢņä▒ņØ┤ Ļ░Ćņן ļåÆņĢśļŗż. Mn@PtņØ┤ ļåÆņØĆ ORR ĒÖ£ņä▒ņØä ļ│┤ņØĖ ņøÉņØĖņØĆ MnņØś ņŚ┤ļ”░ ĻĄ¼ņĪ░ņÖĆ ņŚ░ņä▒ņØ┤ļ®░, Pt ņēśņØś ļæÉĻ╗śņŚÉ ļö░ļØ╝ Mn ņĮöņ¢┤ņØś ĻĄ¼ņĪ░ņĀü Ļ▓░ĒĢ©Ļ│╝ ņøÉņ×É ņČ®ņĀä ļ░ĆļÅäĻ░Ć ļŗ¼ļØ╝ņ¦äļŗż. ņØ┤ņŚÉ ļö░ļØ╝ Pt ņēśļÅä ņĢĢņČĢ, ļ│ĆĒśĢļÉśĻ│Ā Ēæ£ļ®┤ņŚÉ ņ׳ļŖö Pt ņé¼ņØ┤ĒŖĖņØś 5d-ņśżļ╣äĒāłņØ┤ 3D ņāüņŚÉņä£ ņä£ļĪ£ Ļ▓╣ņ╣śĻ▒░ļéś ļŗżļźĖ ļ░®Ē¢źņØä Ļ░ĆņĀĖ ļŗżļźĖ TM@PtņÖĆļŖö ļŗżļźĖ ORR ĒÖ£ņä▒ļÅäļź╝ ļ│┤ņØ╝ ņłś ņ׳ļŗż. ņØ┤ļ¤¼ĒĢ£ ņśüĒ¢źņ£╝ļĪ£ O2ņØś Ļ▓░ĒĢ® ņŚÉļäłņ¦ĆĻ░Ć ļåÆņĢäņĀĖ OH, OOH ņóģņØś Ļ▓░ĒĢ® Ļ░ĢļÅäļź╝ ņ”ØĻ░Ćņŗ£Ēéżņ¦Ć ņĢŖĻ│Ā ņé░ņåī Ļ░äņØś Ļ▓░ĒĢ®ņØä ļüŖņ¢┤ ORR ĒÜ©ņ£©ņØä ļåÆņØĖļŗż. ņĀ£ņØ╝ņøÉļ”¼ Ļ│äņé░ ļ░ĆļÅäļ▓öĒĢ©ņłś ņØ┤ļĪĀ(Density functional theory, DFT)ņŚÉ ĻĖ░ļ░śĒĢśņŚ¼ Pd1-xCux@Pd ņ£äņŚÉ Pt ļŗ©ņØ╝ņĖĄņ£╝ļĪ£ ĻĄ¼ņä▒ļÉ£ ņ┤ēļ¦żņØś ĒÖ£ņä▒, d-band ņŚÉļäłņ¦Ć, ļ░śņØæņżæĻ░äļ¼╝ņØś Ļ▓░ĒĢ® ņŚÉļäłņ¦Ć ļō▒ņØä ļČäņäØĒĢ£ ņŚ░ĻĄ¼ļź╝ ĒåĄĒĢ┤ PdCu ĒĢ®ĻĖł ņĪ░ņä▒ņŚÉ ļö░ļźĖ ORRņŚÉ ļ»Ėņ╣śļŖö ņśüĒ¢źņØ┤ ļ│┤Ļ│ĀļÉśņśĆļŗż [67]. ĻĘĖļ”╝ 7ņŚÉņä£ ļ│┤ņØĖ Ļ▓āĻ│╝ Ļ░ÖņØ┤, d-band center ņŚÉļäłņ¦ĆņÖĆ ņ┤ēļ¦ż Ēæ£ļ®┤ņØś ļ░śņØæņä▒ ņé¼ņØ┤ņŚÉ ņäĀĒśĢņØś Ļ┤ĆĻ│äĻ░Ć ņĪ┤ņ×¼ĒĢ£ļŗżĻ│Ā ņŻ╝ņןĒĢśņśĆļŗż. ņØ┤ļŖö ĒÄśļź┤ļ»Ė ņżĆņ£äņŚÉņä£ ļ©╝ d-band center ņŚÉļäłņ¦ĆņŚÉ ļīĆĒĢ┤ Ēæ£ļ®┤ņØś ļ░śņØæņä▒ņØ┤ ļ¢©ņ¢┤ņ¦äļŗżļŖö Ļ▓āņØä ņØśļ»ĖĒĢ£ļŗż. ņØ┤ņŚÉ ļö░ļØ╝ Pt/Pd/Pd0.875Cu0.125 ņ┤ēļ¦żĻ░Ć ļ░śņØæņä▒ņŚÉ ļīĆĒĢ┤ņä£ ņĄ£ņĀüņØś ņĪ░ņä▒ņØ┤ļØ╝Ļ│Ā ĒĢ£ļŗż.
ņŚŁ ļ¦łņØ┤Ēü¼ļĪ£ ņŚÉļ®ĆņĀ╝ļ▓ĢņØä ņØ┤ņÜ®ĒĢśņŚ¼ ĒÖśņøÉ ĻĘĖļלĒöĮ ņśźņé¼ņØ┤ļō£(rGO)ņŚÉ Pt-Auļź╝ ļŗ┤ņ¦ĆĒĢ£ ņŚ░ĻĄ¼Ļ░Ć ļ│┤Ļ│ĀļÉÉļŗż [65]. Pt/rGOņŚÉ ļ╣äĒĢ┤, Pt-Au/rGOļŖö 50,000 ņé¼ņØ┤Ēü┤ Ēøä ņ┤ēļ¦ż ņä▒ļŖźņØ┤ ļ¦żņÜ░ ņĢłņĀĢĒĢ©ņØä ļ│┤ņśĆļŗż. ĻĘĖļלĒĢĆ ņ¦Ćņ¦Ćņ▓┤ļŖö ņāØņä▒ļÉ£ H2OņØä ņĀ£Ļ▒░ĒĢśļŖöļŹ░ ņĀüĒĢ®ĒĢśņ¦Ć ņĢŖĻ│Ā GDL ĻĄ¼ņĪ░ļŖö Ļ░ĆņŖż ĒÖĢņé░ņŚÉ ņĀüĒĢ®ĒĢśņ¦Ć ņĢŖĻĖ░ ļĢīļ¼ĖņŚÉ ņŖżĒÄśņØ┤ņä£ļź╝ ļÅäņ×ģĒĢśņśĆļŗż. ņŖżĒÄśņØ┤ņä£ņØś ļÅäņ×ģņØĆ GDLņØś ĻĄ¼ņĪ░, ņ┤ēļ¦ż ļČäņé░ļÅä, ņś┤ ņĀĆĒĢŁņØś Ļ░£ņäĀņŚÉ ļÅäņøĆņØä ņŻ╝ņŚłĻ│Ā ņŖżĒÄśņØ┤ņä£ļĪ£ņŹ© CNTņÖĆ Pt-Au/CNTĻ░Ć ņé¼ņÜ®ļÉśņŚłņØä ļĢī Pt-Au/rGO ņ┤ēļ¦żņØś ņĄ£ļīĆ ņĀäļĀź ļ░ĆļÅäĻ░Ć ĻĖēĻ▓®Ē׳ ņ”ØĻ░ĆĒĢśņśĆļŗż.
3. Ļ▓░ ļĪĀ
ļ│Ė ļģ╝ļ¼ĖņŚÉņä£ļŖö ņĄ£ĻĘ╝ ļ░£Ēæ£ļÉ£ ļ░▒ĻĖł ĻĖ░ļ░ś 2ņøÉ ĒĢ®ĻĖł ņ┤ēļ¦żļōżņØä ņåīĻ░£ĒĢśņśĆļŗż. Pt-MņØĆ Pt ņ┤ēļ¦żņŚÉ ļ╣äĒĢ┤ ļ╣äņŖĘĒĢśĻ▒░ļéś ļåÆņØĆ ņä▒ļŖźņØä ļ│┤ņśĆņ£╝ļ®░, ļé┤ĻĄ¼ņä▒Ļ│╝ ņĢłņĀĢņä▒ņØä Ļ░£ņäĀĒĢśņśĆļŗż. Pt-M ņżæ Pt-Ni, Pt-Fe, Pt-CoļŖö ORR Ļ│╝ņĀĢ ņżæ ņé░ņåī ĒØĪĒāłņ░®ņŚÉ ļīĆĒĢ£ ņé░ņåī Ļ▓░ĒĢ® ņŚÉļäłņ¦ĆĻ░Ć ņĀüņĀłĒĢśĻĖ░ ļĢīļ¼ĖņŚÉ ļŗżļźĖ Pt-MņŚÉ ļ╣äĒĢ┤ ņÜ░ņłśĒĢ£ ņ┤ēļ¦ż ņä▒ļŖźņØä ļ│┤ņØĖ Ļ▓āņ£╝ļĪ£ ņÜöņĢĮĒĢĀ ņłś ņ׳ļŗż. ņØ┤ļĪ£ ņØĖĒĢ┤, Pt-Ni, Pt-Fe, Pt-Ni ņ┤ēļ¦ż ĒĢ®ņä▒ņŚÉ Ļ┤ĆĒĢ£ ņŚ░ĻĄ¼Ļ░Ć Ļ░Ćņן ĒÖ£ļ░£Ē׳ ņØ┤ļŻ©ņ¢┤ņĪīņ£╝ļ®░, ĻĘĖ ņżæ Pt-Fe ņ┤ēļ¦żĻ░Ć Ļ░Ćņן ņÜ░ņłśĒĢ£ ņ┤ēļ¦ż ĒÖ£ņä▒ņØä Ļ░Ćņ¦ĆļŖö Ļ▓āņ£╝ļĪ£ ļ│┤ņØĖļŗż. ļ¦ÄņØĆ ņŚ░ĻĄ¼ņŚÉņä£ Pt-MņØś ļ╣äņ£©ņØä ņĪ░ņĀłĒĢśņŚ¼ ņĄ£ņĀüņØś ļ╣äņ£©ņØä ņ░ŠĻĖ░ ņ£äĒĢ┤ ļģĖļĀźĒĢśņśĆļŗż. Ni, Co, FeļŖö Ļ░üĻ░ü PtNi3, PtCo3, PtFe2, PtFe3ņØś ņøÉņ×Éļ╣äļź╝ Ļ░Ćņ¦ł ļĢī ņ┤ēļ¦ż ņä▒ļŖźņØ┤ Ļ░Ćņן ņÜ░ņłśĒĢ£ Ļ▓āņ£╝ļĪ£ ļ│┤ņØ┤ļ®░, ĻĘĖ ņÖĖņØś MņØś Ļ▓ĮņÜ░ Pt:MĻ░Ć PtSn(ņ¦łļ¤ēļ╣ä 1:1) PtPd(ņ¦łļ¤ēļ╣ä 5:95), PtAg(ņ¦łļ¤ēļ╣ä 3:1), Pt3Cr, PtB3 ņØ╝ ļĢī ņä▒ļŖźņØ┤ ņÜ░ņłśĒĢśņśĆļŗż.
Pt ļīĆ M ļ╣äņ£©ņØä ņĪ░ņĀłĒĢśļŖö ļ░®ļ▓Ģ ņÖĖņŚÉļÅä Pt-MņŚÉ ņ╣┤ļ│Ė ļĖöļ×Ö ņÖĖņØś ļŗżņ¢æĒĢ£ ņ¦Ćņ¦Ćņ▓┤(CNT, MWCNT, CNF, CNC, rGO, Gr ļō▒)ļź╝ ņĀüņÜ®ĒĢśĻ▒░ļéś ņĀäĻĄ¼ņ▓┤ ņóģļźś, ņé░ņ▓śļ”¼, ņŚ┤ņ▓śļ”¼ ņĪ░Ļ▒┤ņØä ļŗ¼ļ”¼ĒĢśļŖö ņŚ░ĻĄ¼ļōż, ņ┤ēļ¦żņØś ĻĄ¼ņĪ░ņÖĆ ĒśĢĒā£ļź╝ ņĪ░ņĀłĒĢśņŚ¼ ņä▒ļŖźņØä Ļ░£ņäĀņŗ£ĒéżļŖö ņŚ░ĻĄ¼ļōżņØ┤ ņ׳ņŚłļŗż.